Needleman-Wunsch algorithm
From Wikipedia, the free encyclopedia
The Needleman–Wunsch algorithm performs a global alignment on two sequences (called A and B here). It is commonly used in bioinformatics to align protein or nucleotide sequences. The algorithm was published in 1970 by Saul Needleman and Christian Wunsch[1].
The Needleman–Wunsch algorithm is an example of dynamic programming, and was the first application of dynamic programming to biological sequence comparison.
Scores for aligned characters are specified by a similarity matrix. Here, S(i,j) is the similarity of characters i and j. It uses a linear gap penalty, here called d.
For example, if the similarity matrix was
| - | A | G | C | T |
|---|---|---|---|---|
| A | 10 | -1 | -3 | -4 |
| G | -1 | 7 | -5 | -3 |
| C | -3 | -5 | 9 | 0 |
| T | -4 | -3 | 0 | 8 |
then the alignment:
AGACTAGTTAC CGA---GACGT
with a gap penalty of -5, would have the following score...
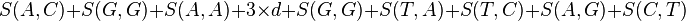

To find the alignment with the highest score, a two-dimensional array (or matrix) is allocated. This matrix is often called the F matrix, and its (i,j)th entry is often denoted Fij There is one column for each character in sequence A, and one row for each character in sequence B. Thus, if we are aligning sequences of sizes n and m, the running time of the algorithm is O(nm) and the amount of memory used is in O(nm). (However, there is a modified version of the algorithm which uses only O(m + n) space, at the cost of a higher running time. This modification is in fact a general technique which applies to many dynamic programming algorithms; this method was introduced in Hirschberg's algorithm for solving the longest common subsequence problem.)
As the algorithm progresses, the Fij will be assigned to be the optimal score for the alignment of the first i characters in A and the first j characters in B. The principle of optimality is then applied as follows.
Basis: F0j = d * j Fi0 = d * i Recursion, based on the principle of optimality: Fij = max(Fi − 1,j − 1 + S(Ai,Bj),Fi,j − 1 + d,Fi − 1,j + d)
The pseudo-code for the algorithm to compute the F matrix therefore looks like this (the sequence indexes start at 1, the F array starts at 0 to include the boundary values defined above):
for i=0 to length(A)
F(i,0) ← d*i
for j=0 to length(B)
F(0,j) ← d*j
for i=1 to length(A)
for j = 1 to length(B)
{
Choice1 ← F(i-1,j-1) + S(A(i), B(j))
Choice2 ← F(i-1, j) + d
Choice3 ← F(i, j-1) + d
F(i,j) ← max(Choice1, Choice2, Choice3)
}
Once the F matrix is computed, note that the bottom right hand corner of the matrix is the maximum score for any alignments. To compute which alignment actually gives this score, you can start from the bottom right cell, and compare the value with the three possible sources(Choice1, Choice2, and Choice3 above) to see which it came from. If Choice1, then A(i) and B(j) are aligned, if Choice2, then A(i) is aligned with a gap, and if Choice3, then B(j) is aligned with a gap. (In general several choices may have the same value, leading to alternative optimal alignments.)
AlignmentA ← ""
AlignmentB ← ""
i ← length(A)
j ← length(B)
while (i > 0 and j > 0)
{
Score ← F(i,j)
ScoreDiag ← F(i - 1, j - 1)
ScoreUp ← F(i, j - 1)
ScoreLeft ← F(i - 1, j)
if (Score == ScoreDiag + S(A(i), B(j)))
{
AlignmentA ← A(i-1) + AlignmentA
AlignmentB ← B(j-1) + AlignmentB
i ← i - 1
j ← j - 1
}
else if (Score == ScoreLeft + d)
{
AlignmentA ← A(i-1) + AlignmentA
AlignmentB ← "-" + AlignmentB
i ← i - 1
}
otherwise (Score == ScoreUp + d)
{
AlignmentA ← "-" + AlignmentA
AlignmentB ← B(j-1) + AlignmentB
j ← j - 1
}
}
while (i > 0)
{
AlignmentA ← A(i-1) + AlignmentA
AlignmentB ← "-" + AlignmentB
i ← i - 1
}
while (j > 0)
{
AlignmentA ← "-" + AlignmentA
AlignmentB ← B(j-1) + AlignmentB
j ← j - 1
}
[edit] References
- ^ Needleman SB, Wunsch CD. (1970). "A general method applicable to the search for similarities in the amino acid sequence of two proteins". J Mol Biol 48 (3): 443-53. doi:. PMID 5420325. http://linkinghub.elsevier.com/retrieve/pii/0022-2836(70)90057-4.
[edit] External links
- Needleman-Wunsch Algorithm as Ruby Code
- Java Implementation of the Needleman-Wunsch Algorithm
- B.A.B.A. — an applet (with source) which visually explains the algorithm.
- A clear explanation of NW and its applications to sequence alignment
- Sequence Alignment Techniques at Technology Blog

