Senescence
From Wikipedia, the free encyclopedia
Senescence is a process induced by evolution into our genetic make up so as we live to our healthiest until our reproductive age and die slowly and gradually thereafter. Senescence encompasses all of the biological processes of a living organism's approaching an advanced age (i.e., the combination of processes of deterioration which follow the period of development of an organism). The word senescence is derived from the Latin word senex, meaning "old man" or "old age" or "advanced in age".
Contents |
[edit] Cellular senescence
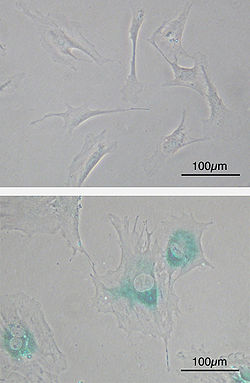
(upper) Primary mouse embryonic fibroblast cells (MEFs) before senescence. Spindle-shaped. (lower) MEFs became senescent after passages. Cells grow larger, flatten shape and expressed senescence-associated β-galactosidase (SABG, blue areas), a marker of cellular senescence.
Cellular senescence is the phenomenon where normal diploid differentiated cells lose the ability to divide, normally after about 50 cell divisions in vitro, some cells become senescent before because of DNA double strand breaks, toxins etc. This phenomenon is also known as "replicative senescence", the "Hayflick phenomenon", or the Hayflick limit in honour of Dr. Leonard Hayflick who was the first to publish this information in 1965. In response to DNA damage (including shortened telomeres) cells either age or self-destruct (apoptosis, programmed cell death) if the damage cannot be repaired. In this 'cellular suicide', the death of one, or more, cells may benefit the organism as a whole. For example, in plants the death of the water-conducting xylem cells (tracheids and vessel elements) allows the cells to function more efficiently and so deliver water to the upper parts of a plant.
[edit] Aging of the whole organism
Organismal senescence is the aging of whole organisms. The term aging has become so commonly equated with senescence that the terms will be used interchangeably in this article. Aging is generally characterized by the declining ability to respond to stress, increasing homeostatic imbalance and increased risk of aging-associated diseases. Because of this, death is the ultimate consequence of aging. Differences in maximum life span among species correspond to different "rates of aging". For example, inherited differences in the rate of aging make a mouse elderly at 3 years and a human elderly at 90 years.[citation needed] These genetic differences affect a variety of physiological processes, including the efficiency of DNA repair, antioxidant enzymes, and rates of free radical production.

Senescence of the organism gives rise to the Gompertz-Makeham law of mortality, which says that mortality rate rises rapidly with age.
Some animals, such as some reptiles and fish, age slowly. Some even exhibit "negative senescence", in which mortality falls with age, in disagreement with the Gompertz-Makeham "law".[1]
Whether replicative senescence (Hayflick limit) plays a causative role in organismal aging is at present an active area of investigation.
[edit] Theories of aging
The process of senescence is complex, and may derive from a variety of different mechanisms and exist for a variety of different reasons. However, senescence is not universal, and scientific evidence suggests that cellular senescence evolved in certain species as a mechanism to prevent the onset of cancer. In a few simple species, senescence is negligible and cannot be detected. All such species have no "post-mitotic" cells; they reduce the effect of damaging free radicals by cell division and dilution. Such species are not immortal, however, as they will eventually fall prey to trauma or disease. Moreover, average lifespans can vary greatly within and between species. This suggests that both genetic and environmental factors contribute to aging.
Traditionally, theories that explain senescence have generally been divided between the programmed and stochastic theories of aging. Programmed theories imply that aging is regulated by biological clocks operating throughout the life span. This regulation would depend on changes in gene expression that affect the systems responsible for maintenance, repair and defense responses. Stochastic theories blame environmental impacts on living organisms that induce cumulative damage at various levels as the cause of aging, examples which range from damage to DNA, damage to tissues and cells by oxygen radicals (widely known as free radicals countered by the even more well known antioxidants), and cross-linking.
Conversely, aging is seen as a progressive failure of homeodynamics (homeostasis) involving genes for the maintenance and repair, stochastic events leading to molecular damage and molecular heterogeneity, and chance events determining the probability of death. Since complex and interacting systems of maintenance and repair comprise the homeodynamic (old term, homeostasis) space of a biological system, aging is considered to be a progressive shrinkage of homeodynamic space mainly due to increased molecular heterogeneity.[citation needed]
[edit] Evolutionary theories
Aging is believed to have evolved because of the increasingly smaller probability of an organism still being alive at older age, due to predation and accidents, both of which may be random and age-invariant. It is thought that strategies which result in a higher reproductive rate at a young age, but shorter overall lifespan, result in a higher lifetime reproductive success and are therefore favoured by natural selection. Essentially, aging is therefore the result of investing resources in reproduction, rather than maintenance of the body (the "Disposable Soma" theory[2]), in light of the fact that accidents, predation and disease will eventually kill the organism no matter how much energy is devoted to repair of the body. Various other, or more specific, theories of aging exist, and are not necessarily mutually exclusive.
The geneticist J. B. S. Haldane wondered why the dominant mutation which causes Huntington's disease remained in the population, and why natural selection had not eliminated it. The onset of this neurological disease is (on average) at age 45 and is invariably fatal within 10–20 years. Haldane assumed, probably reasonably, that in human prehistory, few survived until age 45. Since few were alive at older ages and their contribution to the next generation was therefore small relative to the large cohorts of younger age groups, the force of selection against such late-acting deleterious mutations was correspondingly small. However, if a mutation affected younger individuals, selection against it would be strong. Therefore, late-acting deleterious mutations could accumulate in populations over evolutionary time through genetic drift. This principle has been demonstrated experimentally[citation needed]. And it is these later-acting deleterious mutations which are believed to cause, or perhaps more correctly allow, age-related mortality.
Peter Medawar formalised this observation in his mutation accumulation theory of aging.[3][4] "The force of natural selection weakens with increasing age—even in a theoretically immortal population, provided only that it is exposed to real hazards of mortality. If a genetic disaster... happens late enough in individual life, its consequences may be completely unimportant". The 'real hazards of mortality' are typically predation, disease and accidents. So, even an immortal population, whose fertility does not decline with time, will have fewer individuals alive in older age groups. This is called 'extrinsic mortality.' Young cohorts, not depleted in numbers yet by extrinsic mortality, contribute far more to the next generation than the few remaining older cohorts, so the force of selection against late-acting deleterious mutations, which only affect these few older individuals, is very weak. The mutations may not be selected against, therefore, and may spread over evolutionary time into the population.
The major testable prediction made by this model is that species which have high extrinsic mortality in nature will age more quickly and have shorter intrinsic lifespans. This is borne out among mammals, the most well studied in terms of life history. There is a correlation among mammals between body size and lifespan, such that larger species live longer than smaller species in controlled/optimum conditions, but there are notable exceptions. For instance, many bats and rodents are similarly sized, yet bats live much longer. For instance, the little brown bat, half the size of a mouse, can live 30 years in the wild. A mouse will live 2–3 years even with optimum conditions. The explanation is that bats have fewer predators, so therefore low extrinsic mortality. Thus more individuals survive to later ages so the force of selection against late-acting deleterious mutations is stronger. Fewer late-acting deleterious mutations = slower aging = longer lifespan. Birds are also warm-blooded and similarly sized to many small mammals, yet live often 5–10 times as long. They clearly have fewer predation pressures compared with ground-dwelling mammals. And seabirds, which generally have the fewest predators of all birds, live longest.
Also, when examining the body-size vs. lifespan relationship, predator mammals tend to have longer lifespans than prey animals in a controlled environment such as a zoo or nature reserve. The explanation for the long lifespans of primates (such as humans, monkeys and apes) relative to body size is that their intelligence and often sociality helps them avoid becoming prey. Being a predator, being smart and working together all reduce extrinsic mortality.
Another evolutionary theory of aging was proposed by George C. Williams (Williams 1957)[5] and involves antagonistic pleiotropy. A single gene may affect multiple traits. Some traits that increase fitness early in life may also have negative effects later in life. But because many more individuals are alive at young ages than at old ages, even small positive effects early can be strongly selected for, and large negative effects later may be very weakly selected against. Williams suggested the following example: perhaps a gene codes for calcium deposition in bones which promotes juvenile survival and will therefore be favored by natural selection; however this same gene promotes calcium deposition in the arteries, causing negative effects in old age. Therefore negative effects in old age may reflect the result of natural selection for pleiotropic genes which are beneficial early in life. In this case, fitness is relatively high when Fisher's reproductive value is high and relatively low when Fisher's reproductive value is low.
[edit] Gene regulation
|
|
This article is missing citations or needs footnotes. Please help add inline citations to guard against copyright violations and factual inaccuracies. (July 2008) |
A number of genetic components of aging have been identified using model organisms, ranging from the simple budding yeast Saccharomyces cerevisiae to worms such as Caenorhabditis elegans and fruit flies (Drosophila melanogaster). Study of these organisms has revealed the presence of at least two conserved aging pathways.
One of these pathways involves the gene Sir2, a NAD+-dependent histone deacetylase. In yeast, Sir2 is required for genomic silencing at three loci: the yeast mating loci, the telomeres and the ribosomal DNA (rDNA). In some species of yeast replicative aging may be partially caused by homologous recombination between rDNA repeats; excision of rDNA repeats results in the formation of extrachromosomal rDNA circles (ERCs). These ERCs replicate and preferentially segregate to the mother cell during cell division, and are believed to result in cellular senescence by titrating away (competing for) essential nuclear factors. ERCs have not been observed in other species (nor even all strains of the same yeast species) of yeast (which also display replicative senescence), and ERCs are not believed to contribute to aging in higher organisms such as humans (they have not been shown to accumulate in mammals in a similar manner to yeast). Extrachromosomal circular DNA (eccDNA) has been found in worms, flies and humans. The origin and role of eccDNA in aging, if any, is unknown.
Despite the lack of a connection between circular DNA and aging in higher organisms, extra copies of Sir2 are capable of extending the lifespan of both worms and flies (though in flies, this finding has not been replicated by other investigators, and the activator of Sir2, resveratrol, does not reproducibly increase lifespan in either species[6]). Whether the Sir2 homologues in higher organisms have any role in lifespan is unclear, but the human SIRT1 protein has been demonstrated to deacetylate p53, Ku70, and the forkhead family of transcription factors. SIRT1 can also regulate acetylates such as CBP/p300, and has been shown to deacetylate specific histone residues.
RAS1 and RAS2 also affect aging in yeast and have a human homologue. RAS2 overexpression has been shown to extend lifespan in yeast.
Other genes regulate aging in yeast by increasing the resistance to oxidative stress. Superoxide dismutase, a protein that protects against the effects of mitochondrial free radicals, can extend yeast lifespan in stationary phase when overexpressed.
In higher organisms, aging is likely to be regulated in part through the insulin/IGF-1 pathway. Mutations that affect insulin-like signaling in worms, flies and the growth hormone/IGF1 axis in mice are associated with extended lifespan. In yeast, Sir2 activity is regulated by the nicotinamidase PNC1. PNC1 is transcriptionally upregulated under stressful conditions such as caloric restriction, heat shock, and osmotic shock. By converting nicotinamide to niacin, it removes nicotinamide, which inhibits the activity of Sir2. A nicotinamidase found in humans, known as PBEF, may serve a similar function, and a secreted form of PBEF known as visfatin may help to regulate serum insulin levels. It is not known, however, whether these mechanisms also exist in humans since there are obvious differences in biology between humans and model organisms.
Sir2 activity has been shown to increase under calorie restriction. Due to the lack of available glucose in the cells more NAD+ is available and can activate Sir2. Resveratrol, a polyphenol found in the skin of red grapes, was reported to extend the lifespan of yeast, worms, and flies (the lifespan extension in flies and worms have proved irreproducible by independent investigators[7]). It has been shown to activate Sir2 and therefore mimics the effects of calorie restriction, if one accepts that caloric restriction is indeed depedent on Sir2.
Gene expression is imperfectly controlled, and it is possible that random fluctuations in the expression levels of many genes contribute to the aging process as suggested by a study of such genes in yeast.[8] Individual cells, which are genetically identical, none-the-less can have substantially different responses to outside stimuli, and markedly different lifespans, indicating the epigenetic factors play an important role in gene expression and aging as well as genetic factors.
The following is a list of genes connected to longevity through research[citation needed] on model organisms: the filamentous fungus (Podospora anserina), bakers' yeast (Saccharomyces cerevisiae), the soil roundworm (Caenorhabditis elegans), the fruit fly (Drosophila melanogaster), and the mouse (Mus musculus).
| Podospora | Saccharomyces | Caenorhabditis | Drosophila | Mus | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| grisea | LAG1 | daf-2 | sod1 | Prop-1 | ||||||||
| LAC1 | age-1/daf-23 | cat1 | p66shc (Not independently verified) | pit-1 | Ghr | RAS1 | daf-18 | mth | mclk1 | |||
| RAS2 | akt-1/akt-2 | |||||||||||
| PHB1 | daf-16 | |||||||||||
| PHB2 | daf-12 | |||||||||||
| CDC7 | ctl-1 | |||||||||||
| BUD1 | old-1 | |||||||||||
| RTG2 | spe-26 | |||||||||||
| RPD3 | clk-1 | |||||||||||
| HDA1 | mev-1 | |||||||||||
| SIR2 | ||||||||||||
| aak-2 | ||||||||||||
| SIR4-42 | ||||||||||||
| UTH4 | ||||||||||||
| YGL023 | ||||||||||||
| SGS1 | ||||||||||||
| RAD52 | ||||||||||||
| FOB1 |
[edit] Cellular senescence
As noted above, senescence is not universal, and senescence is not observed in single-celled organisms that reproduce through the process of cellular mitosis.[9][10] Moreover, cellular senescence is not observed in many organisms, including perennial plants, sponges, corals, and lobsters. In those species where cellular senescence is observed, cells eventually become post-mitotic when they can no longer replicate themselves through the process of cellular mitosis -- i.e., cells experience replicative senescence. How and why some cells become post-mitotic in some species has been the subject of much research and speculation, but (as noted above) it is widely believed that cellular senescence evolved as a way to prevent the onset and spread of cancer. Somatic cells that have divided many times will have accumulated DNA mutations and would therefore be in danger of becoming cancerous if cell division continued.
Lately the role of telomeres in cellular senescence has aroused general interest, especially with a view to the possible genetically adverse effects of cloning. The successive shortening of the chromosomal telomeres with each cell cycle is also believed to limit the number of divisions of the cell, thus contributing to aging. There have, on the other hand, also been reports that cloning could alter the shortening of telomeres. Some cells do not age and are therefore described as being "biologically immortal." It is theorized by some that when it is discovered exactly what allows these cells, whether it be the result of telomere lengthening or not, to divide without limit that it will be possible to genetically alter other cells to have the same capability. It is further theorized that it will eventually be possible to genetically engineer all cells in the human body to have this capability by employing gene therapy and thereby stop or reverse aging, effectively making the entire organism potentially immortal.
Cancer cells are usually immortal. This evasion of cellular senescence is the result, in about 85% of tumors, of up-activation of their telomerase genes.[11] This simple observation suggests that reactivation of telomerases in healthy individuals could greatly increase their cancer risk.
Whether cell senescence plays any role in organismal aging is at present unknown, and is an active area of investigation. Mice lacking telomerase do not immediately show accelerated ageing.
[edit] Chemical damage

One of the earliest aging theories was the Rate of Living Hypothesis described by Raymond Pearl in 1928[12](based on earlier work by Max Rubner), which states that fast basal metabolic rate corresponds to short maximum life span (much as a rapidly running machine will experience more damage from wear).
While there may be some validity to the idea that for various types of specific damage detailed below that are by-products of metabolism, all other things being equal, a fast metabolism may reduce lifespan, in general this theory does not adequately explain the differences in lifespan either within, or between, species. Calorically-restricted animals process as much, or more, calories per gram of body mass, as their ad libitum fed counterparts, yet exhibit substantially longer lifespans.[citation needed] . Similarly, metabolic rate is a poor predictor of lifespan for birds, bats and other species which presumably have reduced mortality from predation, and therefore have evolved long lifespans even in the presence of very high metabolic rates[13]. More recently, it was shown that, when modern statistical methods for correcting for the effects of body size and phylogeny are employed, metabolic rate does not correlate with longevity in mammals or birds[14]. (For a critique of the Rate of Living Hypothesis see Living fast, dying when? [15].)
With respect to specific types of chemical damage caused by metabolism, it is suggested that damage to long-lived biopolymers, such as structural proteins or DNA, caused by ubiquitous chemical agents in the body such as oxygen and sugars, are in part responsible for ageing. The damage can include breakage of biopolymer chains, cross-linking of biopolymers, or chemical attachment of unnatural substituents (haptens) to biopolymers.
Under normal aerobic conditions, approximately 4% of the oxygen metabolized by mitochondria is converted to superoxide ion which can subsequently be converted to hydrogen peroxide, hydroxyl radical and eventually other reactive species including other peroxides and singlet oxygen, which can in turn generate free radicals capable of damaging structural proteins and DNA. Certain metal ions found in the body, such as copper and iron, may participate in the process. (In Wilson's disease, a hereditary defect which causes the body to retain copper, some of the symptoms resemble accelerated senescence.) These processes are termed oxidative damage and are linked to the benefits of nutritionally derived polyphenol antioxidants[citation needed].
Sugars such as glucose and fructose can react with certain amino acids such as lysine and arginine and certain DNA bases such as guanine to produce sugar adducts, in a process called glycation. These adducts can further rearrange to form reactive species which can then cross-link the structural proteins or DNA to similar biopolymers or other biomolecules such as non-structural proteins. People with diabetes, who have elevated blood sugar, develop senescence-associated disorders much earlier than the general population, but can delay such disorders by rigorous control of their blood sugar levels. There is evidence that sugar damage is linked to oxidant damage in a process termed glycoxidation.
Free radicals can damage proteins, lipids or DNA. Glycation mainly damages proteins. Damaged proteins and lipids accumulate in lysosomes as lipofuscin. Chemical damage to structural proteins can lead to loss of function; for example, damage to collagen of blood vessel walls can lead to vessel-wall stiffness and thus hypertension, and vessel wall thickening and reactive tissue formation (atherosclerosis); similar processes in the kidney can lead to renal failure. Damage to enzymes reduces cellular functionality. Lipid peroxidation of the inner mitochondrial membrane reduces the electric potential and the ability to generate energy. It is probably no accident that nearly all of the so-called "accelerated aging diseases" are due to defective DNA repair enzymes.
It is believed that the impact of alcohol on aging can be partly explained by alcohol's activation of the HPA axis, which stimulates glucocorticoid secretion; long-term exposure to which produces symptoms of aging.[16]
[edit] Reliability theory
Reliability theory suggests that biological systems start their adult life with a high load of initial damage. Reliability theory is a general theory about systems failure. It allows researchers to predict the age-related failure kinetics for a system of given architecture (reliability structure) and given reliability of its components. Reliability theory predicts that even those systems that are entirely composed of non-aging elements (with a constant failure rate) will nevertheless deteriorate (fail more often) with age, if these systems are redundant in irreplaceable elements. Aging, therefore, is a direct consequence of systems.
Reliability theory also predicts the late-life mortality deceleration with subsequent leveling-off, as well as the late-life mortality plateaus, as an inevitable consequence of redundancy exhaustion at extreme old ages. The theory explains why mortality rates increase exponentially with age (the Gompertz law) in many species, by taking into account the initial flaws (defects) in newly formed systems. It also explains why organisms "prefer" to die according to the Gompertz law, while technical devices usually fail according to the Weibull (power) law. Reliability theory allows to specify conditions when organisms die according to the Weibull distribution: organisms should be relatively free of initial flaws and defects. The theory makes it possible to find a general failure law applicable to all adult and extreme old ages, where the Gompertz and the Weibull laws are just special cases of this more general failure law. The theory explains why relative differences in mortality rates of compared populations (within a given species) vanish with age (compensation law of mortality), and mortality convergence is observed due to the exhaustion of initial differences in redundancy levels.
[edit] Neuro-endocrine-immunological theories
Senescence may also simply be a result of wear and tear overwhelming repair mechanisms. It is also possible that senescence is a mechanism to control the development and spread of cancer; if cells have built-in limits to how many times they can replicate, they must somehow overcome this before they can spread indefinitely.
[edit] Miscellaneous
Recently, early senescence has been alleged to be a possible unintended outcome of early cloning experiments. Most notably, the issue was raised in the case of Dolly the sheep, following her death from a contagious lung disease. The claim that Dolly's early death involved premature senescence has been vigorously contested (e.g. by Kerry Lynn Macintosh in her book, Illegal Beings: Human Clones and the Law), and Dolly's creator, Dr. Ian Wilmut has expressed the view that her illness and death were probably unrelated to the fact that she was a clone.
A set of rare hereditary (genetic) disorders, each called progeria, has been known for some time. Sufferers exhibit symptoms resembling accelerated aging, including wrinkled skin. The cause of Hutchinson–Gilford progeria syndrome was reported in the journal Nature in May 2003. This report suggests that DNA damage, not oxidative stress, is the cause of this form of accelerated ageing.
[edit] See also
| Look up senescence in Wiktionary, the free dictionary. |
- Advanced adult
- Ageing
- Ageing brain
- Calorie restriction
- Evolution of ageing
- Strategies for Engineered Negligible Senescence (SENS)
- Fisher's reproductive value
- Life extension
- List of life extension-related topics
- Maximum life span
- Mitohormesis
- Plant senescence
- Progeria
- Real death
- SAGE KE
- Sub-lethal damage
[edit] References
- ^ New Scientist 195.2616 (August 11, 2007): p36(4).[1]
- ^ Kirkwood, T.B.L. 1977. Evolution of ageing. Nature, 270: 301-304. [2]
- ^ Medawar, P. B., 1946 Old age and natural death. Mod. 1:30–56.
- ^ Medawar, Peter B. (1952). An Unsolved Problem of Biology. London: H. K. Lewis.
- ^ Williams, G. C., 1957 Pleiotropy, natural selection and the evolution of senescence. Evolution 11:398-411.
- ^ Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech Ageing Dev. 2007 Oct;128(10):546-52.
- ^ Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech Ageing Dev. 2007 Oct;128(10):546-52.
- ^ Ryley J, Pereira-Smith OM (2006). "Microfluidics device for single cell gene expression analysis in Saccharomyces cerevisiae". Yeast 23 (14-15): 1065–73. PMID 17083143.
- ^ Gavrilov, L. A., Gavrilova, N. S., 2001 The reliability theory of ageing and longevity. Journal of Theoretical Biology 213(4): 527-545. PMID 11742523
- ^ F. Yaghmaie, O. Saeed, S.A. Garan, M.A. Voelker, A.M. Gouw, W. Freitag, H. Sternberg and P.S. Timiras "Age-dependent loss of insulin-like growth factor-1 receptor immunoreactive cells in the supraoptic hypothalamus is reduced in calorically restricted mice". International Journal of Developmental Neuroscience, Vol. 24, Issue 7, 2006, pp. 431-436
- ^ Hanahan D, Weinberg RA (2000). "The hallmarks of cancer". Cell 100 (1): 57–70. doi:. PMID 10647931.
- ^ Pearl, Raymond (1928). The Rate of Living, Being an Account of Some Experimental Studies on the Biology of Life Duration. New York: Alfred A. Knopf.
- ^ Brunet-Rossinni AK, Austad SN (2004). "Aging studies on bats: a review". BIOGERONTOLOGY 5 (4): 211–222. doi:. PMID 15314271.
- ^ de Magalhaes JP, Costa J, Church GM. An analysis of the relationship between metabolism, developmental schedules, and longevity using phylogenetic independent contrasts. J Gerontol A Biol Sci Med Sci. 2007 Feb;62(2):149-60 PMID 17339640
- ^ Speakman JR, Selman C, McLaren JS, Harper EJ (2002). "Living fast, dying when? The link between aging and energetics". THE JOURNAL OF NUTRITION 132 (6, Supplement 2): 1583S–1597S. PMID 12042467. http://jn.nutrition.org/cgi/content/full/132/6/1583S.
- ^ http://pubs.niaaa.nih.gov/publications/arh23-4/272-283.pdf
[edit] External links
- AgeLab (MIT).
- Ageing Research Centre (ARC)
- American Academy of Anti-Ageing Medicine
- American Federation for Ageing Research
- Gerontology Research Group Site also has the official tables of known supercentenarians.
- Leibniz Institute for Age Research - Fritz Lipmann Institute (FLI)
- Longevity Meme (Longevity Activism)
- Longevity Science
- Mechanisms of Ageing
- Ouroboros Anti-Ageing Research News Written by scientists for scientists
- 54 Scientists' Open Letter on Ageing Research
- senescence.info Educational resource on the science of ageing.
- Greenwood Research on Anti-Aging Medicine and Longevity
- himanshusenescence[3]

