Mitochondrion
From Wikipedia, the free encyclopedia
In addition to supplying cellular energy, mitochondria are involved in a range of other processes, such as signaling, cellular differentiation, cell death, as well as the control of the cell cycle and cell growth.[1] Mitochondria have been implicated in several human diseases, including mitochondrial disorders[2] and cardiac dysfunction,[3] and may play a role in the aging process. The word mitochondrion comes from the Greek μίτος or mitos, thread + χονδρίον or khondrion, granule. Several characteristics make mitochondria unique. The number of mitochondria in a cell varies widely by organism and tissue type. Many cells have only a single mitochondrion, whereas others can contain several thousand mitochondria.[4][5] The organelle is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, the intermembrane space, the inner membrane, and the cristae and matrix. Mitochondrial proteins vary depending on the tissues and species. In human, 615 distinct types of proteins were identified from cardiac mitochondria;[6] whereas in murinae (rats), 940 proteins encoded by distinct genes were reported.[7] The mitochondrial proteome is thought to be dynamically regulated.[8] Although most of a cell's DNA is contained in the cell nucleus, the mitochondrion has its own independent genome. Further, its DNA shows substantial similarity to bacterial genomes.[9]
Contents |
[edit] Structure
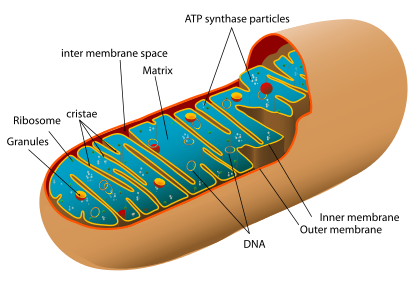
A mitochondrion contains outer and inner membranes composed of phospholipid bilayers and proteins.[4] The two membranes, however, have different properties. Because of this double-membraned organization, there are five distinct compartments within the mitochondrion. There is the outer mitochondrial membrane, the intermembrane space (the space between the outer and inner membranes), the inner mitochondrial membrane, the cristae space (formed by infoldings of the inner membrane), and the matrix (space within the inner membrane).
[edit] Outer membrane
The outer mitochondrial membrane, which encloses the entire organelle, has a protein-to-phospholipid ratio similar to that of the eukaryotic plasma membrane (about 1:1 by weight). It contains large numbers of integral proteins called porins. These porins form channels that allow molecules 5000 Daltons or less in molecular weight to freely diffuse from one side of the membrane to the other.[4] Larger proteins can also enter the mitochondrion if a signaling sequence at their N-terminus binds to a large multisubunit protein called translocase of the outer membrane, which then actively moves them across the membrane.[10] Disruption of the outer membrane permits proteins in the intermembrane space to leak into the cytosol, leading to certain cell death.[11]
[edit] Intermembrane space
The intermembrane space is basically the space between the outer membrane and the inner membrane. Because the outer membrane is freely permeable to small molecules, the concentrations of small molecules such as ions and sugars in the intermembrane space is the same as the cytosol.[4] However, as large proteins must have a specific signaling sequence to be transported across the outer membrane, the protein composition of this space is different than the protein composition of the cytosol. One protein that is localized to the intermembrane space in this way is cytochrome c.[11]
[edit] Inner membrane
The inner mitochondrial membrane contains proteins with four types of functions:[4]
- Those that perform the redox reactions of oxidative phosphorylation
- ATP synthase, which generates ATP in the matrix
- Specific transport proteins that regulate metabolite passage into and out of the matrix
- Protein import machinery.
It contains more than 100 different polypeptides, and has a very high protein-to-phospholipid ratio (more than 3:1 by weight, which is about 1 protein for 15 phospholipids). The inner membrane is home to around 1/5 of the total protein in a mitochondrion.[4] In addition, the inner membrane is rich in an unusual phospholipid, cardiolipin. This phospholipid was originally discovered in beef hearts in 1942, and is usually characteristic of mitochondrial and bacterial plasma membranes.[12] Cardiolipin contains four fatty acids rather than two and may help to make the inner membrane impermeable.[4] Unlike the outer membrane, the inner membrane does not contain porins and is highly impermeable to all molecules. Almost all ions and molecules require special membrane transporters to enter or exit the matrix. Proteins are ferried into the matrix via the translocase of the inner membrane (TIM) complex or via Oxa1.[10] In addition, there is a membrane potential across the inner membrane formed by the action of the enzymes of the electron transport chain.
[edit] Cristae

The inner mitochondrial membrane is compartmentalized into numerous cristae, which expand the surface area of the inner mitochondrial membrane, enhancing its ability to produce ATP. These are not simple random folds but rather invaginations of the inner membrane, which can affect overall chemiosmotic function.[13] In typical liver mitochondria, for example, the surface area, including cristae, is about five times that of the outer membrane. Mitochondria of cells that have greater demand for ATP, such as muscle cells, contain more cristae than typical liver mitochondria.[4]
[edit] Matrix
The matrix is the space enclosed by the inner membrane. It contains about 2/3 of the total protein in a mitochondrion.[4] The matrix is important in the production of ATP with the aid of the ATP synthase contained in the inner membrane. The matrix contains a highly-concentrated mixture of hundreds of enzymes, special mitochondrial ribosomes, tRNA, and several copies of the mitochondrial DNA genome. Of the enzymes, the major functions include oxidation of pyruvate and fatty acids, and the citric acid cycle.[4]
Mitochondria have their own genetic material, and the machinery to manufacture their own RNAs and proteins (see: protein biosynthesis). A published human mitochondrial DNA sequence revealed 16,569 base pairs encoding 37 total genes: 22 tRNA, 2 rRNA, and 13 peptide genes.[14] The 13 mitochondrial peptides in humans are integrated into the inner mitochondrial membrane, along with proteins encoded by genes that reside in the host cell's nucleus.
[edit] Organization and distribution
Mitochondria are found in nearly all eukaryotes. They vary in number and location according to cell type. Substantial numbers of mitochondria are in the liver, with about 1000–2000 mitochondria per cell making up 1/5th of the cell volume.[4] The mitochondria can be found nestled between myofibrils of muscle or wrapped around the sperm flagellum.[4] Often they form a complex 3D branching network inside the cell with the cytoskeleton. The association with the cytoskeleton determines mitochondrial shape, which can affect the function as well.[15] Recent evidence suggests vimentin, one of the components of the cytoskeleton, is critical to the association with the cytoskeleton.[16]
[edit] Function
The most prominent roles of the mitochondrion are its production of ATP and regulation of cellular metabolism.[5] The central set of reactions involved in ATP production are collectively known as the citric acid cycle, or the Krebs Cycle. However, the mitochondrion has many other functions in addition to the production of ATP.
[edit] Energy conversion
A dominant role for the mitochondria is the production of ATP, as reflected by the large number of proteins in the inner membrane for this task. This is done by oxidizing the major products of glucose, pyruvate, and NADH, which are produced in the cytosol.[5] This process of cellular respiration, also known as aerobic respiration, is dependent on the presence of oxygen. When oxygen is limited, the glycolytic products will be metabolized by anaerobic respiration, a process that is independent of the mitochondria.[5] The production of ATP from glucose has an approximately 13-fold higher yield during aerobic respiration compared to anaerobic respiration.[17] Recently it has been shown that plant mitochondria can produce a limited amount of ATP without oxygen by using the alternate substrate nitrite.[18]
[edit] Pyruvate: the citric acid cycle
Each pyruvate molecule produced by glycolysis is actively transported across the inner mitochondrial membrane, and into the matrix where it is oxidized and combined with coenzyme A to form CO2, acetyl-CoA, and NADH.[5]
The acetyl-CoA is the primary substrate to enter the citric acid cycle, also known as the tricarboxylic acid (TCA) cycle or Krebs cycle. The enzymes of the citric acid cycle are located in the mitochondrial matrix, with the exception of succinate dehydrogenase, which is bound to the inner mitochondrial membrane as part of Complex II.[19] The citric acid cycle oxidizes the acetyl-CoA to carbon dioxide, and, in the process, produces reduced cofactors (three molecules of NADH and one molecule of FADH2) that are a source of electrons for the electron transport chain, and a molecule of GTP (that is readily converted to an ATP).[5]
[edit] NADH and FADH2: the electron transport chain
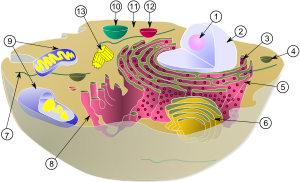
(1) Nucleolus
(2) nucleus
(3) Ribosomes
(4) vesicle
(5) Rough endoplasmic reticulum (ER)
(6) Golgi apparatus
(7) Cytoskeleton
(8) Smooth ER
(9) mitochondria
(10) Vacuole
(11) Cytoplasm
(12) Lysosome
(13) Centrioles within Centrosome
The redox energy from NADH and FADH2 is transferred to oxygen (O2) in several steps via the electron transport chain. These energy-rich molecules are produced within the matrix via the citric acid cycle but are also produced in the cytoplasm by glycolysis. Reducing equivalents from the cytoplasm can be imported via the malate-aspartate shuttle system of antiporter proteins or feed into the electron transport chain using a glycerol phosphate shuttle.[5] Protein complexes in the inner membrane (NADH dehydrogenase, cytochrome c reductase, and cytochrome c oxidase) perform the transfer and the incremental release of energy is used to pump protons (H+) into the intermembrane space. This process is efficient, but a small percentage of electrons may prematurely reduce oxygen, forming reactive oxygen species such as superoxide.[5] This can cause oxidative stress in the mitochondria and may contribute to the decline in mitochondrial function associated with the aging process.[20]
As the proton concentration increases in the intermembrane space, a strong electrochemical gradient is established across the inner membrane. The protons can return to the matrix through the ATP synthase complex, and their potential energy is used to synthesize ATP from ADP and inorganic phosphate (Pi).[5] This process is called chemiosmosis, and was first described by Peter Mitchell[21][22] who was awarded the 1978 Nobel Prize in Chemistry for his work. Later, part of the 1997 Nobel Prize in Chemistry was awarded to Paul D. Boyer and John E. Walker for their clarification of the working mechanism of ATP synthase.[23]
[edit] Heat production
Under certain conditions, protons can re-enter the mitochondrial matrix without contributing to ATP synthesis. This process is known as proton leak or mitochondrial uncoupling and is due to the facilitated diffusion of protons into the matrix. The process results in the unharnessed potential energy of the proton electrochemical gradient being released as heat.[5] The process is mediated by a proton channel called thermogenin, or UCP1.[24] Thermogenin is a 33kDa protein first discovered in 1973.[25] Thermogenin is primarily found in brown adipose tissue, or brown fat, and is responsible for non-shivering thermogenesis. Brown adipose tissue is found in mammals, and is at its highest levels in early life and in hibernating animals. In humans, brown adipose tissue is present at birth and decreases with age.[24]
[edit] Storage of calcium ions
The concentrations of free calcium in the cell can regulate an array of reactions and is important for signal transduction in the cell. Mitochondria can transiently store calcium, a contributing process for the cell's homeostasis of calcium.[26] In fact, their ability to rapidly take in calcium for later release makes them very good "cytosolic buffers" for calcium.[27] The endoplasmic reticulum (ER) is the most significant storage site of calcium, and there is a significant interplay between the mitochondrion and ER with regard to calcium.[28] The calcium is taken up into the matrix by a calcium uniporter on the inner mitochondrial membrane.[29] It is primarily driven by the mitochondrial membrane potential.[26] Release of this calcium back into the cell's interior can occur via a sodium-calcium exchange protein or via "calcium-induced-calcium-release" pathways.[29] This can initiate calcium spikes or calcium waves with large changes in the membrane potential. These can activate a series of second messenger system proteins that can coordinate processes such as neurotransmitter release in nerve cells and release of hormones in endocrine cells.
[edit] Additional functions
Mitochondria play a central role in many other metabolic tasks, such as:
- Regulation of the membrane potential[5]
- Apoptosis-programmed cell death[30]
- Calcium signaling (including calcium-evoked apoptosis)[31]
- Cellular proliferation regulation[32]
- Regulation of cellular metabolism[32]
- Certain heme synthesis reactions[33] (see also: porphyrin)
- Steroid synthesis.[27]
Some mitochondrial functions are performed only in specific types of cells. For example, mitochondria in liver cells contain enzymes that allow them to detoxify ammonia, a waste product of protein metabolism. A mutation in the genes regulating any of these functions can result in mitochondrial diseases.
[edit] Origin
Mitochondria have many features in common with prokaryotes. As a result, they are believed to be originally derived from endosymbiotic prokaryotes.
A mitochondrion contains DNA, which is organized as several copies of a single, circular chromosome. This mitochondrial chromosome contains genes for redox proteins such as those of the respiratory chain. The CoRR Hypothesis proposes that this Co-location is required for Redox Regulation. The mitochondrial genome also codes for some RNAs of ribosomes, and the twenty-two tRNA's necessary for the translation of messenger RNAs into protein. The circular structure is also found in prokaryotes, and the similarity is extended by the fact that mitochondrial DNA is organized with a variant genetic code similar to that of Proteobacteria.[34] This suggests that their ancestor, the so-called proto-mitochondrion, was a member of the Proteobacteria.[34] In particular, the proto-mitochondrion was probably related to the rickettsia.[35] However, the exact relationship of the ancestor of mitochondria to the alpha-proteobacteria and whether the mitochondria was formed at the same time or after the nucleus, remains controversial.[36]
The ribosomes coded for by the mitochondrial DNA are similar to those from bacteria in size and structure.[37] They closely resemble the bacterial 70S ribosome and not the 80S cytoplasmic ribosomes which are coded for by nuclear DNA.
The endosymbiotic relationship of mitochondria with their host cells was popularized by Lynn Margulis.[38] The endosymbiotic hypothesis suggests that mitochondria descended from bacteria that somehow survived endocytosis by another cell, and became incorporated into the cytoplasm. The ability of these bacteria to conduct respiration in host cells that had relied on glycolysis and fermentation would have provided a considerable evolutionary advantage. In a similar manner, host cells with symbiotic bacteria capable of photosynthesis would also have had an advantage. The incorporation of symbiotes would have increased the number of environments in which the cells could survive. This symbiotic relationship probably developed 1.7[39]-2[40] billion years ago.
A few groups of unicellular eukaryotes lack mitochondria: the microsporidians, metamonads, and archamoebae.[41] These groups appear as the most primitive eukaryotes on phylogenetic trees constructed using rRNA information, suggesting that they appeared before the origin of mitochondria. However, this is now known to be an artifact of long-branch attraction – they are derived groups and retain genes or organelles derived from mitochondria (e.g., mitosomes and hydrogenosomes).[42]
[edit] Genome
The human mitochondrial genome is a circular DNA molecule of about 16 kilobases.[43] It encodes 37 genes: 13 for subunits of respiratory complexes I, III, IV and V, 22 for mitochondrial tRNA (for the 20 standard amino acids, plus an extra gene for leucine and serine), and 2 for rRNA.[43] One mitochondrion can contain two to ten copies of its DNA.[44]
As in prokaryotes, there is a very high proportion of coding DNA and an absence of repeats. Mitochondrial genes are transcribed as multigenic transcripts, which are cleaved and polyadenylated to yield mature mRNAs. Not all proteins necessary for mitochondrial function are encoded by the mitochondrial genome; most are coded by genes in the cell nucleus and the corresponding proteins imported into the mitochondrion.[45] The exact number of genes encoded by the nucleus and the mitochondrial genome differs between species. In general, mitochondrial genomes are circular, although exceptions have been reported.[46] Also, in general, mitochondrial DNA lacks introns, as is the case in the human mitochondrial genome;[45] however, introns have been observed in some eukaryotic mitochondrial DNA,[47] such as that of yeast[48] and protists,[49] including Dictyostelium discoideum.[50]
In animals the mitochondrial genome is typically a single circular chromosome that is approximately 16-kb long and has 37 genes. The genes while highly conserved may vary in location. Curiously this pattern is not found in the human body louse (Pediculus humanus). Instead this mitochondrial genome is arranged in 18 minicircular chromosomes each of which is 3-4 kb long and has one to three genes.[51] This pattern is also found in other sucking lice but not in chewing lice. Recombination has been shown to occur between the minichromosomes. The reason for this difference is not known.
While slight variations on the standard code had been predicted earlier,[52] none was discovered until 1979, when researchers studying human mitochondrial genes determined that they used an alternative code.[53] Many slight variants have been discovered since,[54] including various alternative mitochondrial codes.[55] Further, the AUA, AUC, and AUU codons are all allowable start codons.
| Organism | Codon | Standard | Novel |
|---|---|---|---|
| Mammalian | AGA, AGG | Arginine | Stop codon |
| AUA | Isoleucine | Methionine | |
| UGA | Stop codon | Tryptophan | |
| Invertebrates | AGA, AGG | Arginine | Serine |
| AUA | Isoleucine | Methionine | |
| UGA | Stop codon | Tryptophan | |
| Yeast | AUA | Isoleucine | Methionine |
| UGA | Stop codon | Tryptophan | |
| CUA | Leucine | Threonine |
Some of these differences should be regarded as pseudo-changes in the genetic code due to the phenomenon of RNA editing, which is common in mitochondria. In higher plants, it was thought that CGG encoded for tryptophan and not arginine; however, the codon in the processed RNA was discovered to be the UGG codon, consistent with the universal genetic code for tryptophan.[56] Of note, the arthropod mitochondrial genetic code has undergone parallel evolution within a phylum, with some organisms uniquely translating AGG to lysine.[57]
Mitochondrial genomes have far fewer genes than the bacteria from which they are thought to be descended. Although some have been lost altogether, many have been transferred to the nucleus, such as the respiratory complex II protein subunits.[43] This is thought to be relatively common over evolutionary time. A few organisms, such as the Cryptosporidium, actually have mitochondria that lack any DNA, presumably because all their genes have been lost or transferred.[58] In Cryptosporidium, the mitochondria have an altered ATP generation system that renders the parasite resistant to many classical mitochondrial inhibitors such as cyanide, azide, and atovaquone.[58]
[edit] Replication and inheritance
Mitochondria divide by binary fission similar to bacterial cell division; unlike bacteria, however, mitochondria can also fuse with other mitochondria.[43][59]. The regulation of this division differs between eukaryotes. In many single-celled eukaryotes, their growth and division is linked to the cell cycle. For example, a single mitochondrion may divide synchronously with the nucleus. This division and segregation process must be tightly controlled so that each daughter cell receives at least one mitochondrion. In other eukaryotes (in humans for example), mitochondria may replicate their DNA and divide mainly in response to the energy needs of the cell, rather than in phase with the cell cycle. When the energy needs of a cell are high, mitochondria grow and divide. When the energy use is low, mitochondria are destroyed or become inactive. In such examples, and in contrast to the situation in many single celled eukaryotes, mitochondria are apparently randomly distributed to the daughter cells during the division of the cytoplasm.
An individual's mitochondrial genes are not inherited by the same mechanism as nuclear genes. At fertilization of an egg cell by a sperm, the egg nucleus and sperm nucleus each contribute equally to the genetic makeup of the zygote nucleus. In contrast, the mitochondria, and therefore the mitochondrial DNA, usually comes from the egg only. The sperm's mitochondria enter the egg but do not contribute genetic information to the embryo.[60] Instead, paternal mitochondria are marked with ubiquitin to select them for later destruction inside the embryo.[61] The egg cell contains relatively few mitochondria, but it is these mitochondria that survive and divide to populate the cells of the adult organism. Mitochondria are, therefore, in most cases inherited down the female line, known as maternal inheritance. This mode is seen in most organisms including all animals. However, mitochondria in some species can sometimes be inherited paternally. This is the norm among certain coniferous plants, although not in pine trees and yew trees.[62] It has also been suggested that it occurs at a very low level in humans.[63]
Uniparental inheritance leads to little opportunity for genetic recombination between different lineages of mitochondria, although a single mitochondrion can contain 2–10 copies of its DNA.[44] For this reason, mitochondrial DNA usually is thought to reproduce by binary fission. What recombination does take place maintains genetic integrity rather than maintaining diversity. However, there are studies showing evidence of recombination in mitochondrial DNA. It is clear that the enzymes necessary for recombination are present in mammalian cells.[64] Further, evidence suggests that animal mitochondria can undergo recombination.[65] The data are a bit more controversial in humans, although indirect evidence of recombination exists.[66][67] If recombination does not occur, the whole mitochondrial DNA sequence represents a single haplotype, which makes it useful for studying the evolutionary history of populations.
[edit] Population genetic studies
The near-absence of genetic recombination in mitochondrial DNA makes it a useful source of information for scientists involved in population genetics and evolutionary biology.[68] Because all the mitochondrial DNA is inherited as a single unit, or haplotype, the relationships between mitochondrial DNA from different individuals can be represented as a gene tree. Patterns in these gene trees can be used to infer the evolutionary history of populations. The classic example of this is in human evolutionary genetics, where the molecular clock can be used to provide a recent date for mitochondrial Eve.[69][70] This is often interpreted as strong support for a recent modern human expansion out of Africa.[71] Another human example is the sequencing of mitochondrial DNA from Neanderthal bones. The relatively-large evolutionary distance between the mitochondrial DNA sequences of Neanderthals and living humans has been interpreted as evidence for lack of interbreeding between Neanderthals and anatomically-modern humans.[72]
However, mitochondrial DNA reflects the history of only females in a population and so may not represent the history of the population as a whole. This can be partially overcome by the use of paternal genetic sequences, such as the non-recombining region of the Y-chromosome.[71] In a broader sense, only studies that also include nuclear DNA can provide a comprehensive evolutionary history of a population.[73]
[edit] Dysfunction and disease
[edit] Mitochondrial diseases
With their central place in cell metabolism, damage - and subsequent dysfunction - in mitochondria is an important factor in a wide range of human diseases. Mitochondrial disorders often present as neurological disorders, but can manifest as myopathy, diabetes, multiple endocrinopathy, or a variety of other systemic manifestations.[74] Diseases caused by mutation in the mtDNA include Kearns-Sayre syndrome, MELAS syndrome and Leber's hereditary optic neuropathy.[75] In the vast majority of cases, these diseases are transmitted by a female to her children, as the zygote derives its mitochondria and hence its mtDNA from the ovum. Diseases such as Kearns-Sayre syndrome, Pearson's syndrome, and progressive external ophthalmoplegia are thought to be due to large-scale mtDNA rearrangements, whereas other diseases such as MELAS syndrome, Leber's hereditary optic neuropathy, myoclonic epilepsy with ragged red fibers (MERRF), and others are due to point mutations in mtDNA.[74]
In other diseases, defects in nuclear genes lead to dysfunction of mitochondrial proteins. This is the case in Friedreich's ataxia, hereditary spastic paraplegia, and Wilson's disease.[76] These diseases are inherited in a dominance relationship, as applies to most other genetic diseases. A variety of disorders can be caused by nuclear mutations of oxidative phosphorylation enzymes, such as coenzyme Q10 deficiency and Barth syndrome.[74] Environmental influences may also interact with hereditary predispositions and cause mitochondrial disease. For example, there may be a link between pesticide exposure and the later onset of Parkinson's disease.[77][78]
Other pathologies with etiology involving mitochondrial dysfunction include schizophrenia, bipolar disorder, dementia, Alzheimer's disease, Parkinson's disease, epilepsy, stroke, cardiovascular disease, retinitis pigmentosa, and diabetes mellitus.[79][80] A common thread thought to link these seemingly-unrelated conditions is cellular damage causing oxidative stress. How exactly mitochondrial dysfunction fits into the etiology of these pathologies is yet to be elucidated.[citation needed]
[edit] Possible relationships to aging
Given the role of mitochondria as the cell's powerhouse, there may be some leakage of the high-energy electrons in the respiratory chain to form reactive oxygen species. This can result in significant oxidative stress in the mitochondria with high mutation rates of mitochondrial DNA.[81] A vicious cycle is thought to occur, as oxidative stress leads to mitochondrial DNA mutations, which can lead to enzymatic abnormalities and further oxidative stress. A number of changes occur to mitochondria during the aging process.[82] Tissues from elderly patients show a decrease in enzymatic activity of the proteins of the respiratory chain.[83] Large deletions in the mitochondrial genome can lead to high levels of oxidative stress and neuronal death in Parkinson's disease.[84] Hypothesized links between aging and oxidative stress are not new and were proposed over 50 years ago;[85] however, there is much debate over whether mitochondrial changes are causes of aging or merely characteristics of aging. One notable study in mice demonstrated shortened lifespan but no increase in reactive oxygen species despite increasing mitochondrial DNA mutations, suggesting that mitochondrial DNA mutations can cause lifespan shortening by other mechanisms.[86] As a result, the exact relationships between mitochondria, oxidative stress, and aging have not yet been settled.
[edit] References
- ^ McBride HM, Neuspiel M, Wasiak S (2006). "Mitochondria: more than just a powerhouse". Curr. Biol. 16 (14): R551. doi:. PMID 16860735.
- ^ Gardner A, Boles RG (2005). "Is a "Mitochondrial Psychiatry" in the Future? A Review". Curr. Psychiatry Review 1 (3): 255–271. doi:.
- ^ Lesnefsky EJ, et al. (2001). "Mitochondrial dysfuntion in cardiac disease ischemia-reperfusion, aging and heart failure". J. Mol. Cell. Cardiol. 33 (6): 1065–1089. doi:.
- ^ a b c d e f g h i j k l m Alberts, Bruce; Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, Peter Walter (1994). Molecular Biology of the Cell. New York: Garland Publishing Inc.. ISBN 0815332181.
- ^ a b c d e f g h i j k Voet, Donald; Judith G. Voet, Charlotte W. Pratt (2006). Fundamentals of Biochemistry, 2nd Edition. John Wiley and Sons, Inc.. pp. 547. ISBN 0471214957.
- ^ Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS (2003 March). "Characterization of the human heart mitochondrial proteome". Nat Biotechnol. 21 (3): 281–6. doi:. PMID 12592411.
- ^ Zhang J, Li X, Mueller M, Wang Y, Zong C, Deng N, Vondriska TM, Liem DA, Yang J, Korge P, Honda H, Weiss JN, Apweiler R, Ping P (2008). "Systematic characterization of the murine mitochondrial proteome using functionally validated cardiac mitochondira". Proteomics 8 (8): 1564–1575. doi:. PMID 18348319.
- ^ Zhang J, Liem DA, Mueller M, Wang Y, Zong C, Deng N, Vondriska TM, Yang J, Korge P, Drews O, Maclellan WR, Honda H, Weiss JN, Apweiler R, Ping P (2008). "Altered Proteome Biology of Cardiac Mitochondria Under Stress Conditions". J. Proteome Res 7: 2204. doi:. PMID 18484766.
- ^ Andersson SG, Karlberg O, Canbäck B, Kurland CG (January 2003). "On the origin of mitochondria: a genomics perspective". Philos. Trans. R. Soc. Lond., B, Biol. Sci. 358 (1429): 165–77; discussion 177–9. doi:. PMID 12594925.
- ^ a b Herrmann JM, Neupert W (2000 April). "Protein transport into mitochondria". Curr Opin Microbiol 3 (2): 210–214. doi:.
- ^ a b Chipuk JE, Bouchier-Hayes L, Green DR (2006). "Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario". Cell Death and Differentiation. 13: 1396–1402. doi:.
- ^ McMillin JB, Dowhan W (2002 December). "Cardiolipin and apoptosis". Biochim. Et Biophys. Acta. 1585: 97–107. doi:. PMID 12531542.
- ^ Mannella CA (2006). "Structure and dynamics of the mitochondrial inner membrane cristae". Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 1763 (5–6): 542–548. doi:. PMID 16730811.
- ^ Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, et al. (1981-04-09). "Sequence and organization of the human mitochondrial genome". Nature. 410 (5806): 141. doi:.
- ^ Rappaport L, Oliviero P, Samuel JL (1998). "Cytoskeleton and mitochondrial morphology and function". Mol and Cell Biochem. 184: 101–105. doi:.
- ^ Tang HL, Lung HL, Wu KC, Le AP, Tang HM, Fung MC (2007). "Vimentin supports mitochondrial morphology and organization". Biochemical J 410: 141. doi:. PMID 17983357.
- ^ Rich PR (2003). "The molecular machinery of Keilin's respiratory chain". Biochem. Soc. Trans. 31 (Pt 6): 1095–105. doi:. PMID 14641005. http://www.biochemsoctrans.org/bst/031/1095/bst0311095.htm.
- ^ Stoimenova M, Igamberdiev AU, Gupta KJ, Hill RD (July 2007). "Nitrite-driven anaerobic ATP synthesis in barley and rice root mitochondria". Planta 226 (2): 465–74. doi:. PMID 17333252.
- ^ King A, Selak MA, Gottlieb E (2006). "Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer". Oncogene. 25: 4675–4682. doi:.
- ^ Huang, K.; K. G. Manton (2004). "The role of oxidative damage in mitochondria during aging: A review". Frontiers in Bioscience 9: 1100–1117. doi:.
- ^ Mitchell P, Moyle J (1967-01-14). "Chemiosmotic hypothesis of oxidative phosphorylation". Nature. 213 (5072): 137–9. doi:.
- ^ Mitchell P (1967-06-24). "Proton current flow in mitochondrial systems". Nature. 25 (5095): 1327–8. doi:. PMID 6056845.
- ^ Nobel Foundation. "Chemistry 1997". http://nobelprize.org/nobel_prizes/chemistry/laureates/1997/. Retrieved on 2007-12-16.
- ^ a b Mozo J, Emre Y, Bouillaud F, Ricquier D, Criscuolo F (2005 November). "Thermoregulation: What Role for UCPs in Mammals and Birds?". Bioscience Reports. 25: 227–249. doi:.
- ^ Nicholls DG, Lindberg O (1973). "Brown-adipose-tissue mitochondria. The influence of albumin and nucleotides on passive ion permeabilities". Eur. J. Biochem. 37: R551. doi:. PMID 4777251.
- ^ a b Siegel GJ, Agranoff BW, Fisher SK, Albers RW, Uhler MD, ed (1999). Basic Neurochemistry (6 ed.). Lippincott Williams & Wilkins. ISBN 0-397-51820-X.
- ^ a b Rossier MF (2006). "T channels and steroid biosynthesis: in search of a link with mitochondria". Cell Calcium. 40 (2): 155–64. doi:. PMID 16759697.
- ^ Pizzo P, Pozzan T (2007 October). "Mitochondria–endoplasmic reticulum choreography: structure and signaling dynamics". Trends Cell Bio. 17 (10): 511–517. doi:. PMID 17851078.
- ^ a b Miller RJ (1998). "Mitochondria – the kraken wakes!". Trends in Neurosci. 21 (3): 95–97 doi=10.1016/S0166–2236(97)01206–X. doi:.
- ^ Green DR (1998 September). "Apoptotic pathways: the roads to ruin". Cell. 94 (6): 695–8. doi:. PMID 9753316.
- ^ Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M (2006). "Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis". Cell Calcium 40: 553. doi:. PMID 17074387.
- ^ a b McBride HM, Neuspiel M, Wasiak S (2006 July). "Mitochondria: more than just a powerhouse". Curr Biol. 16 (14): R551–60. doi:. PMID 16860735.
- ^ Oh-hama T (1997 August). "Evolutionary consideration on 5-aminolevulinate synthase in nature". Orig Life Evol Biosph. 27 (4): 405–12. doi:. PMID 9249985.
- ^ a b Futuyma DJ (2005). "On Darwin's Shoulders". Natural History 114 (9): 64–68.
- ^ Emelyanov VV (2003). "Mitochondrial connection to the origin of the eukaryotic cell". Eu J Biochem. 270 (8): 1599–1618. doi:.
- ^ Gray MW, Burger G, Lang BF (March 1999). "Mitochondrial evolution". Science (journal) 283 (5407): 1476–81. PMID 10066161.
- ^ O'Brien TW (2003 September). "Properties of human mitochondrial ribosomes". IUBMB Life. 55 (9): 505–13. doi:.
- ^ Lynn Sagan (1967). "On the origin of mitosing cells". J Theor Bio. 14 (3): 255–274. doi:. PMID 11541392.
- ^ Emelyanov VV (2001). "Rickettsiaceae, rickettsia-like endosymbionts, and the origin of mitochondria". Biosci. Rep. 21: 1–17. doi:. PMID 11508688.
- ^ Feng D-F, Cho G, Doolittle RF (1997). "Determining divergence times with a protein clock: update and reevaluation". Proc. Natl Acad. Sci. 94: 13028–13033. doi:. PMID 9371794.
- ^ Cavalier-Smith T (1991). "Archamoebae: the ancestral eukaryotes?". Biosystems. 25: 1241. doi:. PMID 1854912.
- ^ Henze K, Martin W (2003). "Evolutionary biology: essence of mitochondria". Nature 426 (6963): 127–8. doi:. PMID 14614484.
- ^ a b c d Chan DC (2006-06-30). "Mitochondria: Dynamic Organelles in Disease, Aging, and Development". Cell 125 (7): 1241–1252. doi:. PMID 16814712.
- ^ a b Wiesner RJ, Ruegg JC, Morano I (1992). "Counting target molecules by exponential polymerase chain reaction, copy number of mitochondrial DNA in rat tissues". Biochim Biophys Acta 183: 553–559. PMID 1550563.
- ^ a b Anderson S, Bankier AT, Barrell BG, de-Bruijn MHL, Coulson AR, et al. (1981). "Sequence and organization of the human mitochondrial genome". Nature 290: 427–465. doi:.
- ^ Fukuhara H, Sor F, Drissi R, Dinouël N, Miyakawa I, Rousset, and Viola AM (1993). "Linear mitochondrial DNAs of yeasts: frequency of occurrence and general features". Mol Cell Biol. 13 (4): 2309–2314. PMID 8455612.
- ^ Bernardi G (1978). "Intervening sequences in the mitochondrial genome". Nature. 276 (5688): 558–559. doi:. PMID 214710.
- ^ Hebbar SK, Belcher SM, Perlman PS (1992 April). "A maturase-encoding group IIA intron of yeast mitochondria self-splices in vitro". Nucleic Acids Res. 20 (7): 1747–54. doi:. PMID 1579468.
- ^ Gray MW, Lang BF, Cedergren R, Golding GB, Lemieux C, Sankoff D, et al (1998). "Genome structure and gene content in protist mitochondrial DNAs". Nucl Acids Res. 26 (4): 865–878. doi:. PMID 9461442.
- ^ Gray MW, Lang BF, Burger G (2004). "Mitochondria of protists". Ann Rev of Genetics. 38: 477–524. doi:. PMID 15568984.
- ^ Shao R, Kirkness EF, Barker SC (March 2009). "The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus". Genome Res.. doi:. PMID 19336451.
- ^ Crick, F. H. C. and Orgel, L. E. (1973) "Directed panspermia." Icarus 19:341-346. p. 344: "It is a little surprising that organisms with somewhat different codes do not coexist." (Further discussion at [1])
- ^ Barrell BG, Bankier AT, Drouin J (1979). "A different genetic code in human mitochondria". Nature. 282: 189–194. doi:.
- ^ NCBI: "The Genetic Codes", Compiled by Andrzej (Anjay) Elzanowski and Jim Ostell
- ^ Jukes TH, Osawa S (1990-12-01). "The genetic code in mitochondria and chloroplasts". Experientia. 46 (11–12): 1117–26. doi:. PMID 2253709.
- ^ Hiesel R, Wissinger B, Schuster W, Brennicke A (2006). "RNA editing in plant mitochondria". Science. 246 (4937): 1632–4. doi:. PMID 2480644.
- ^ Abascal F, Posada D, Knight RD, Zardoya R (2006). "Parallel Evolution of the Genetic Code in Arthropod Mitochondrial Genomes". PLoS Biology. 4 (5): 0711–0718. doi:. PMID 16620150.
- ^ a b Henriquez FL, Richards TA, Roberts F, McLeod R, Roberts CW (2005 February). "The unusual mitochondrial compartment of Cryptosporidium parvum". Trends Parasitol. 21 (2): 68–74. doi:. PMID 15664529.
- ^ Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM (1998 October). "Mitochondrial Fusion in Yeast Requires the Transmembrane GTPase Fzo1p". J. Cell. Bio. 143 (2): 359–373. doi:. PMID 9786948.
- ^ Kimball, J.W. (2006) "Sexual Reproduction in Humans: Copulation and Fertilization," Kimball's Biology Pages (based on Biology, 6th ed., 1996)]
- ^ Sutovsky, P., et. al (1999). "Ubiquitin tag for sperm mitochondria". Nature 402: 371–372. doi:. Discussed in Science News.
- ^ Mogensen HL (1996). "The Hows and Whys of Cytoplasmic Inheritance in Seed Plants". American Journal of Botany 83: 247. doi:.
- ^ Johns, D. R. (2003). "Paternal transmission of mitochondrial DNA is (fortunately) rare". Annals of Neurology 54: 422–4. doi:. PMID 14520651.
- ^ Thyagarajan B, Padua RA, Campbell C (1996). "Mammalian mitochondria possess homologous DNA recombination activity". J. Biol. Chem. 271 (44): 27536–27543. doi:. PMID 8910339.
- ^ Lunt DB, Hyman BC (15 May 1997). "Animal mitochondrial DNA recombination". Nature 387: 247. doi:. PMID 9153388.
- ^ Eyre-Walker A, Smith NH, Maynard Smith J (1999-03-07). "How clonal are human mitochondria?". Proc. Royal Soc. Biol. Sci. (Series B) 266 (1418): 477–483. doi:. PMID 10189711.
- ^ Awadalla P, Eyre-Walker A, Maynard Smith J (24 December 1999). "Linkage Disequilibrium and Recombination in Hominid Mitochondrial DNA". Science. 286 (5449): 2524–2525. doi:. PMID 10617471.
- ^ Castro JA, Picornell A, Ramon M (1998). "Mitochondrial DNA: a tool for populational genetics studies". Int Microbiol. 1 (4): 327–32. PMID 10943382.
- ^ Cann RL, Stoneking M, Wilson AC (1987 January). "Mitochondrial DNA and human evolution". Nature. 325: 31–36. doi:.
- ^ Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (2006). "Harvesting the fruit of the human mtDNA tree". Trends Genet. 22 (6): 339–45. doi:. PMID 16678300.
- ^ a b Garrigan D, Hammer MF (2006). "Reconstructing human origins in the genomic era". Nat. Rev. Genet. 7 (9): 669–80. doi:. PMID 16921345.
- ^ Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S (1997). "Neandertal DNA sequences and the origin of modern humans". Cell 90 (1): 19–30. doi:. PMID 9230299.
- ^ Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB (1997 April). "Archaic African and Asian lineages in the genetic ancestry of modern humans". Am J Hum Genet. 60 (4): 772–89. PMID 9106523.
- ^ a b c Zeviani M, Di Donato S (2004). "Mitochondrial disorders". Brain. 127: 2153–2172. doi:. PMID 15358637.
- ^ Taylor RW, Turnbull DM (2005). "Mitochondrial DNA mutations in human disease". Nat. Rev. Genet. 6 (5): 389–402. doi:. PMID 15861210.
- ^ Chinnery PF, Schon EA (2003). "Mitochondria". J. Neurol. Neurosurg. Psychiatr. 74 (9): 1188–99. doi:. PMID 12933917.
- ^ Sherer TB, Betarbet R, Greenamyre JT (2002). "Environment, mitochondria, and Parkinson's disease". The Neuroscientist. 8 (3): 192–7. doi:. PMID 12061498.
- ^ Gomez C, Bandez MJ, Navarro A (2007). "Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome". Front. Biosci. 12: 1079–93. doi:. PMID 17127363.
- ^ Schapira AH (2006). "Mitochondrial disease". Lancet 368 (9529): 70–82. doi:. PMID 16815381.
- ^ Pieczenik SR, Neustadt J (2007). "Mitochondrial dysfunction and molecular pathways of disease". Exp. Mol. Pathol. 83 (1): 84–92. doi:. PMID 17239370.
- ^ Richter C, Park J, Ames BN (1988 September). "Normal Oxidative Damage to Mitochondrial and Nuclear DNA is Extensive". PNAS 85 (17): 6465–6467. doi:. PMID 3413108.
- ^ "Mitochondria and Aging.". http://www.circuitblue.com/biogerontology/mito.shtml.
- ^ Boffoli D, Scacco SC, Vergari R, Solarino G, Santacroce G, Papa S (1994). "Decline with age of the respiratory chain activity in human skeletal muscle". Biochim. Biophys. Acta. 1226: 73–82. PMID 8155742.
- ^ Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006). "High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease". Nat Gen. 38: 515–517. doi:. PMID 16604074.
- ^ Harman D (1956). "Aging: a theory based on free radical and radiation chemistry". J. Gerontol. 11: 298–300. PMID 13332224.
- ^ Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG (2005). "Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production". PNAS. 102 (50): 17993–8. doi:. PMID 16332961.
[edit] See also
| Wikimedia Commons has media related to: Mitochondrion |
- Anti-mitochondrial antibodies
- Bioenergetics
- CoRR Hypothesis
- Human mitochondrial genetics
- Mitochondrial permeability transition pore
- Oncocyte
- Oncocytoma
- Protofection
- Submitochondrial particle
[edit] External links
- Mitochondria Atlas at University of Mainz
- Mitochondria Research Portal at mitochondrial.net
- Mitochondria: Architecture dictates function at cytochemistry.net
- Mitochondria links at University of Alabama
- MIP Mitochondrial Physiology Society
- Mitochondrion Reconstructed by Electron Tomography at San Diego State University
- Video Clip of Rat-liver Mitochondrion from Cryo-electron Tomography at wadsworth.org
- 3D structures of proteins from inner mitochondrial membrane at University of Michigan
- 3D structures of proteins associated with outer mitochondrial membrane at University of Michigan
- Mitochondria meet Art
|
|||||||||||||||||
This article contains material from the Science Primer published by the NCBI, which, as a U.S. government publication, is in the public domain.

