Protein structure
From Wikipedia, the free encyclopedia
|
|
This article may require cleanup to meet Wikipedia's quality standards. Please improve this article if you can. (February 2009) |
|
|
This article needs additional citations for verification. Please help improve this article by adding reliable references (ideally, using inline citations). Unsourced material may be challenged and removed. (March 2009) |
Proteins are an important class of biological macromolecules present in all biological organisms, made up of such elements as carbon,hydrogen, nitrogen, oxygen, and sulphur. All proteins are polymers of amino acids. The polymers, also known as polypeptides consist of a sequence of 20 different L-α-amino acids, also referred to as residues. For chains under 40 residues the term peptide is frequently used instead of protein. To be able to perform their biological function, proteins fold into one, or more, specific spatial conformations, driven by a number of noncovalent interactions such as hydrogen bonding, ionic interactions, Van der Waals forces and hydrophobic packing. In order to understand the functions of proteins at a molecular level, it is often necessary to determine the three dimensional structure of proteins. This is the topic of the scientific field of structural biology, that employs techniques such as X-ray crystallography or NMR spectroscopy, to determine the structure of proteins.
A number of residues are necessary to perform a particular biochemical function, and around 40-50 residues appears to be the lower limit for a functional domain size. Protein sizes range from this lower limit to several thousand residues in multi-functional or structural proteins. However, the current estimate for the average protein length is around 300 residues.[1] Very large aggregates can be formed from protein subunits, for example many thousand actin molecules assemble into a microfilament.
[edit] Levels of protein structure

Biochemistry refers to four distinct aspects of a protein's structure:
- Primary structure - the amino acid sequence of the peptide chains.
- Secondary structure - highly regular sub-structures (alpha helix and strands of beta sheet) which are locally defined, meaning that there can be many different secondary motifs present in one single protein molecule.
- Tertiary structure - three-dimensional structure of a single protein molecule; a spatial arrangement of the secondary structures. It also describes the completely folded and compacted polypeptide chain.
- Quaternary structure - complex of several protein molecules or polypeptide chains, usually called protein subunits in this context, which function as part of the larger assembly or protein complex.
In addition to these levels of structure, a protein may shift between several similar structures in performing its biological function. This process is also reversible. In the context of these functional rearrangements, these tertiary or quaternary structures are usually referred to as chemical conformation, and transitions between them are called conformational changes.
The primary structure is held together by covalent or peptide bonds, which are made during the process of protein biosynthesis or translation. These peptide bonds provide rigidity to the protein. The two ends of the amino acid chain are referred to as the C-terminal end or carboxyl terminus (C-terminus) and the N-terminal end or amino terminus (N-terminus) based on the nature of the free group on each extremity.
The various types of secondary structure are defined by their patterns of hydrogen bonds between the main-chain peptide groups. However, these hydrogen bonds are generally not stable by themselves, since the water-amide hydrogen bond is generally more favorable than the amide-amide hydrogen bond. Thus, secondary structure is stable only when the local concentration of water is sufficiently low, e.g., in the molten globule or fully folded states.
Similarly, the formation of molten globules and tertiary structure is driven mainly by structurally non-specific interactions, such as the rough propensities of the amino acids and hydrophobic interactions. However, the tertiary structure is fixed only when the parts of a protein domain are locked into place by structurally specific interactions, such as ionic interactions (salt bridges), hydrogen bonds and the tight packing of side chains. The tertiary structure of extracellular proteins can also be stabilized by disulfide bonds, which reduce the entropy of the unfolded state; disulfide bonds are extremely rare in cytosolic proteins, since the cytosol is generally a reducing environment.
[edit] Structure of the amino acids

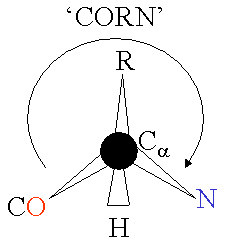
An α-amino acid consists of a part that is present in all the amino acid types, and a side chain that is unique to each type of residue. The Cα atom is bound to 4 different atoms: a hydrogen atom (the H is omitted in the diagram), an amino group nitrogen, a carboxyl group carbon, and a side chain carbon specific for this type of amino acid. An exception from this rule is proline, where the hydrogen atom is replaced by a bond to the side chain. Because the carbon atom is bound to four different groups it is chiral, however only one of the isomers occur in biological proteins. Glycine however, is not chiral since its side chain is a hydrogen atom. A simple mnemonic for correct L-form is "CORN": when the Cα atom is viewed with the H in front, the residues read "CO-R-N" in a clockwise direction.
The side chain determines the chemical properties of the α-amino acid and may be any one of the 20 different side chains:
[edit] Primary structure of proteins
The primary structure of peptides and proteins refers to the linear number and order of the amino acids present. The convention for the designation of the order of amino acids is that the N-terminal end (i.e. the end bearing the residue with the free α-amino group) is to the left (and the number 1 amino acid) and the C-terminal end (i.e. the end with the residue containing a free α-carboxyl group) is to the right.
The proposal that proteins were linear chains of α-amino acids was made nearly simultaneously by two scientists at the same conference in 1902, the 74th meeting of the Society of German Scientists and Physicians, held in Karlsbad. Franz Hofmeister made the proposal in the morning, based on his observations of the biuret reaction in proteins. Hofmeister was followed a few hours later by Emil Fischer, who had amased a wealth of chemical details supporting the peptide-bond model. For completeness, the proposal that proteins contained amide linkages was made as early as 1882 by the French chemist E. Grimaux.
Despite these data and later evidence that proteolytically digested proteins yielded only oligopeptides, the idea that proteins were linear, unbranched polymers of amino acids was not accepted immediately. Some well-respected scientists such as William Astbury doubted that covalent bonds were strong enough to hold such long molecules together; they feared that thermal agitations would shake such long molecules asunder. Hermann Staudinger faced similar prejudices in the 1920s when he argued that rubber was composed of macromolecules.
Thus, several alternative hypotheses arose. The colloidal protein hypothesis stated that proteins were colloidal assemblies of smaller molecules. This hypothesis was disproven in the 1920s by ultracentrifugation measurements by The Svedberg that showed that proteins had a well-defined, reproducible molecular weight and by electrophoretic measurements by Arne Tiselius that indicated that proteins were single molecules. A second hypothesis, the cyclol hypothesis advanced by Dorothy Wrinch, proposed that the linear polypeptide underwent a chemical cyclol rearrangement C=O + HN C(OH)-N that crosslinked its backbone amide groups, forming a two-dimensional fabric. Other primary structures of proteins were proposed by various researchers, such as the diketopiperazine model of Emil Abderhalden and the pyrrol/piperidine model of Troensegaard in 1942. Although never given much credence, these alternative models were finally disproven when Frederick Sanger successfully sequenced insulin and by the crystallographic determination of myoglobin and hemoglobin by Max Perutz and John Kendrew.
The primary structure of a biological polymer to a large extent determines the three-dimensional shape known as the tertiary structure, but nucleic acid and protein folding are so complex that knowing the primary structure often doesn't help either to deduce the shape or to predict localized secondary structure, such as the formation of loops or helices. However, knowing the structure of a similar homologous sequence (for example a member of the same protein family) can unambiguously identify the tertiary structure of the given sequence. Sequence families are often determined by sequence clustering, and structural genomics projects aim to produce a set of representative structures to cover the sequence space of possible non-redundant sequences.
[edit] Secondary structure in proteins
The ordered array of amino acids in a protein confer regular conformational forms upon that protein. These conformations constitute the secondary structures of a protein. In general proteins fold into two broad classes of structure termed, globular proteins and fibrous proteins. Globular proteins are compactly folded and coiled, whereas, fibrous proteins are more filamentous or elongated. It is the partial double-bond character of the peptide bond that defines the conformations a polypeptide chain may assume. Within a single protein different regions of the polypeptide chain may assume different conformations determined by the primary sequence of the amino acids.
[edit] The α-Helix
The α-helix is a common secondary structure encountered in proteins of the globular class. About 35% of all amino acids in proteins are in α-helices, but in individual protein molecules this number ranges from 0 to 80%. The formation of the α-helix is spontaneous and is stabilized by H-bonding between amide nitrogens and carbonyl carbons of peptide bonds spaced four residues apart. This orientation of H-bonding produces a helical coiling of the peptide backbone such that the R-groups lie on the exterior of the helix and perpendicular to its axis. The average length of α-helices is 10.5 amino acids, but the range is from 4 to several dozens.
Not all amino acids favor the formation of the α-helix due to steric constraints of the R-groups. Amino acids such as A, D, E, I, L and M favor the formation of α-helices, whereas, G and P favor disruption of the helix. This is particularly true for P since it is a pyrrolidine based imino acid (HN=) whose structure significantly restricts movement about the peptide bond in which it is present, thereby, interfering with extension of the helix. The disruption of the helix is important as it introduces additional folding of the polypeptide backbone to allow the formation of globular proteins.
[edit] β-sheets
Whereas an α-helix is composed of a single linear array of helically disposed amino acids, β-sheets are composed of 2 or more different regions of stretches of at least 3-5 amino acids (average 4.5 amino acids). The folding and alignment of stretches of the polypeptide backbone aside one another to form β-sheets is stabilized by H-bonding between amide nitrogens and carbonyl carbons. However, the H-bonding residues are present in adjacently opposed stretches of the polypetide backbone as opposed to a linearly contiguous region of the backbone in the α-helix. β-sheets are said to be pleated. This is due to positioning of the α-carbons of the peptide bond which alternates above and below the plane of the sheet. β-sheets are either parallel or antiparallel. In parallel sheets adjacent peptide chains proceed in the same direction (i.e. the direction of N-terminal to C-terminal ends is the same), whereas, in antiparallel sheets adjacent chains are aligned in opposite directions. Most anti-parallel β-sheets are from stretches adjacent in the sequence, connected by just a short loop. On the other hand, parallel β-sheets are always separated by a longer stretch of amino acids. β-sheets can be depicted in ball and stick format or as ribbons in certain protein formats.
Ball and Stick Representation of a β-SheetRibbon Depiction of β-Sheet
[edit] Super-Secondary Structure
Many proteins contain an ordered organization of several adjacent elements of secondary structures that form distinct, commonly observed structural motifs larger than individual secondary structures but smaller than domains or subunits. They are often hypothesized to act as early steps in the process of protein folding. Examples include β-hairpins, helix hairpins, right-handed β-α-β loops, and the helix-turn-helix motifs of bacterial proteins that regulate transcription.
[edit] Tertiary Structure of Proteins
Tertiary structure refers to the complete three-dimensional structure of the polypeptide units of a given protein. Included in this description is the spatial relationship of different secondary structures to one another within a polypeptide chain and how these secondary structures themselves fold into the three-dimensional form of the protein. Secondary structures of proteins often constitute distinct domains. Therefore, tertiary structure also describes the relationship of different domains to one another within a protein. The interactions of different domains is governed by several forces: These include hydrogen bonding, hydrophobic interactions, electrostatic interactions, van der Waals forces and covalent bonding with use of disulfide bridges.
[edit] Quaternary Structure
Many proteins contain 2 or more different polypeptide chains that are held in association by the same non-covalent forces that stabilize the tertiary structures of proteins. Proteins with multiple polypetide chains are oligomeric proteins. The structure formed by monomer-monomer interaction in an oligomeric protein is known as quaternary structure.
Oligomeric proteins can be composed of multiple identical polypeptide chains or multiple distinct polypeptide chains. Proteins with identical subunits are termed homo-oligomers. Proteins containing several distinct polypeptide chains are termed hetero-oligomers.
Hemoglobin, the oxygen carrying protein of the blood, contains two α and two β subunits arranged with a quaternary structure in the form, α2β2. Hemoglobin is, therefore, a hetero-oligomeric protein(I. Shahid et al., 2008). LUCAS
[edit] Forces Controlling Protein Structure
[edit] Hydrogen Bonding
Polypeptides contain numerous proton donors and acceptors both in their backbone and in the R-groups of the amino acids. The environment in which proteins are found also contains ample H-bond donors and acceptors of the water molecule. H-bonding, therefore, occurs not only within and between polypeptide chains but with the surrounding aqueous medium.
[edit] Hydrophobic Forces
Proteins are composed of amino acids that contain either hydrophilic or hydrophobic R-groups. It is the nature of the interaction of the different R-groups with the aqueous environment that plays the major role in shaping protein structure. The spontaneous folded state of globular proteins is a reflection of a balance between the opposing energetics of H-bonding between hydrophilic R-groups and the aqueous environment and the repulsion from the aqueous environment by the hydrophobic R-groups. The hydrophobicity of certain amino acid R-groups tends to drive them away from the exterior of proteins and into the interior. This driving force restricts the available conformations into which a protein may fold.
[edit] Electrostatic Forces
Electrostatic forces are mainly of three types; charge-charge, charge-dipole and dipole-dipole. Typical charge-charge interactions that favor protein folding are those between oppositely charged R-groups such as K or R and D or E. A substantial component of the energy involved in protein folding is charge-dipole interactions. This refers to the interaction of ionized R-groups of amino acids with the dipole of the water molecule. The slight dipole moment that exist in the polar R-groups of amino acid also influences their interaction with water. It is, therefore, understandable that the majority of the amino acids found on the exterior surfaces of globular proteins contain charged or polar R-groups.
[edit] van der Waals Forces
There are both attractive and repulsive van der Waals forces that control protein folding. Attractive van der Waals forces involve the interactions among induced dipoles that arise from fluctuations in the charge densities that occur between adjacent uncharged non-bonded atoms. Repulsive van der Waals forces involve the interactions that occur when uncharged non-bonded atoms come very close together but do not induce dipoles. The repulsion is the result of the electron-electron repulsion that occurs as two clouds of electrons begin to overlap. Although van der Waals forces are extremely weak, relative to other forces governing conformation, it is the huge number of such interactions that occur in large protein molecules that make them significant to the folding of proteins.
[edit] Complex Protein Structures
Proteins also are found to be covalently conjugated with carbohydrates. These modifications occur following the synthesis (translation) of proteins and are, therefore, termed post-translational modifications. These forms of modification impart specialized functions upon the resultant proteins. Proteins covalently associated with carbohydrates are termed glycoproteins. Glycoproteins are of two classes, N-linked and O-linked, referring to the site of covalent attachment of the sugar moieties. N-linked sugars are attached to the amide nitrogen of the R-group of asparagine; O-linked sugars are attached to the hydroxyl groups of either serine or threonine and occasionally to the hydroxyl group of the modified amino acid, hydroxylysine.
There are extremely important glycoproteins found on the surface of erythrocytes. It is the variability in the composition of the carbohydrate portions of many glycoproteins and glycolipids of erythrocytes that determines blood group specificities. There are at least 100 blood group determinants, most of which are due to carbohydrate differences. The most common blood groups, A, B, and O, are specified by the activity of specific gene products whose activities are to incorporate distinct sugar groups onto RBC membrane glycoshpingolipids as well as secreted glycoproteins.
Structural complexes involving protein associated with lipid via noncovalent interactions are termed lipoproteins. The distinct roles of lipoproteins are described on the linked page. Their major function in the body is to aid in the storage transport of lipid and cholesterol.
[edit] Amino-Terminal Sequence Determination
Prior to sequencing peptides it is necessary to eliminate disulfide bonds within peptides and between peptides. Several different chemical reactions can be used in order to permit separation of peptide strands and prevent protein conformations that are dependent upon disulfide bonds. The most common treatments are to use either 2-mercaptoethanol or dithiothreitol (DTT). Both of these chemicals reduce disulfide bonds. To prevent reformation of the disulfide bonds the peptides are treated with iodoacetic acid in order to alkylate the free sulfhydryls.
There are three major chemical techniques for sequencing peptides and proteins from the N-terminus. These are the Sanger, Dansyl chloride and Edman techniques.
- Sanger's Reagent
- This sequencing technique utilizes the compound, 2,4-dinitrofluorobenzene (DNF) which reacts with the N-terminal residue under alkaline conditions. The derivatized amino acid can be hydrolyzed and will be labeled with a dinitrobenzene group that imparts a yellow color to the amino acid. Separation of the modified amino acids (DNP-derivative) by electrophoresis and comparison with the migration of DNP-derivative standards allows for the identification of the N-terminal amino acid.
- Dansyl chloride
- Like DNF, dansyl chloride reacts with the N-terminal residue under alkaline conditions. Analysis of the modified amino acids is carried out similarly to the Sanger method except that the dansylated amino acids are detected by fluorescence. This imparts a higher sensitivity into this technique over that of the Sanger method.
- Edman degradation
- The utility of the Edman degradation technique is that it allows for additional amino acid sequence to be obtained from the N-terminus inward. Using this method it is possible to obtain the entire sequence of peptides. This method utilizes phenylisothiocyanate to react with the N-terminal residue under alkaline conditions. The resultant phenylthiocarbamyl derivatized amino acid is hydrolyzed in anhydrous acid. The hydrolysis reaction results in a rearrangement of the released N-terminal residue to a phenylthiohydantoin derivative. As in the Sanger and Dansyl chloride methods, the N-terminal residue is tagged with an identifiable marker, however, the added advantage of the Edman process is that the remainder of the peptide is intact. The entire sequence of reactions can be repeated over and over to obtain the sequences of the peptide. This process has subsequently been automated to allow rapid and efficient sequencing of even extremely small quantities of peptide.
| Name (Residue) | 3-letter code |
Single code |
Relative abundance (%) E.C. |
MW | pK | VdW volume (ų) |
Charged, Polar, Hydrophobic, Neutral |
|---|---|---|---|---|---|---|---|
| Alanine | ALA | A | 13.0 | 71 | 67 | H | |
| Arginine | ARG | R | 5.3 | 157 | 12.5 | 148 | C+ |
| Asparagine | ASN | N | 9.9 | 114 | 96 | P | |
| Aspartate | ASP | D | 9.9 | 114 | 3.9 | 91 | C- |
| Cysteine | CYS | C | 1.8 | 103 | 86 | P | |
| Glutamate | GLU | E | 10.8 | 128 | 4.3 | 109 | C- |
| Glutamine | GLN | Q | 10.8 | 128 | 114 | P | |
| Glycine | GLY | G | 7.8 | 57 | 48 | N | |
| Histidine | HIS | H | 0.7 | 137 | 6.0 | 118 | P,C+ |
| Isoleucine | ILE | I | 4.4 | 113 | 124 | H | |
| Leucine | LEU | L | 7.8 | 113 | 124 | H | |
| Lysine | LYS | K | 7.0 | 129 | 10.5 | 135 | C+ |
| Methionine | MET | M | 3.8 | 131 | 124 | H | |
| Phenylalanine | PHE | F | 3.3 | 147 | 135 | H | |
| Proline | PRO | P | 4.6 | 97 | 90 | H | |
| Serine | SER | S | 6.0 | 87 | 73 | P | |
| Threonine | THR | T | 4.6 | 101 | 93 | P | |
| Tryptophan | TRP | W | 1.0 | 186 | 163 | P | |
| Tyrosine | TYR | Y | 2.2 | 163 | 10.1 | 141 | P |
| Valine | VAL | V | 6.0 | 99 | 105 | H |
The 20 naturally occurring amino acids can be divided into several groups based on their chemical proporties. Important factors are charge, hydrophobicity/hydrophilicity, size and functional groups. The nature of the interaction of the different side chains with the aqueous environment plays a major role in molding protein structure. Hydrophobic side chains tends to be buried in the middle of the protein, whereas hydrophilic side chains are exposed to the solvent.
Examples of hydrophobic residues are: Leucine, isoleucine, phenylalanine, and valine, and to a lesser extent tyrosine, alanine and tryptophan. The charge of the side chains plays an important role in protein structures, since ion bonding can stabilize proteins structures, and an unpaired charge in the middle of a protein can disrupt structures. Charged residues are strongly hydrophilic, and are usually found on the out side of proteins. Positively charged side chains are found in lysine and arginine, and in some cases in histidine. Negative charges are found in glutamate and aspartate. The rest of the amino acids have smaller generally hydrophilic side chains with various functional groups. Serine and threonine have hydroxylgroups, and aspargine and glutamine have amide groups. Some amino acids have special properties such as cysteine, that can form covalent disulfide bonds to other cysteines, proline that is cyclical, and glycine that is small, and more flexible than the other amino acids.
[edit] The peptide bond
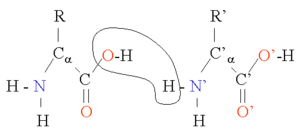

Two amino acids can be combined in a condensation reaction. By repeating this reaction, long chains of residues (amino acids in a peptide bond) can be generated. This reaction is catalysed by the ribosome in a process known as translation. The peptide bond is in fact planar due to the delocalization of the electrons from the double bond. The rigid peptide dihedral angle, ω (the bond between C1 and N) is always close to 180 degrees. The dihedral angles phi φ (the bond between N and Cα) and psi ψ (the bond between Cα and C1) can have a certain range of possible values. These angles are the degrees of freedom of a protein, they control the protein's three dimensional structure. They are restrained by geometry to allowed ranges typical for particular secondary structure elements, and represented in a Ramachandran plot. A few important bond lengths are given in the table below.
| Peptide bond | Average length | Single bond | Average length | Hydrogen bond | Average (±30) |
| Ca - C | 153 pm | C - C | 154 pm | O-H --- O-H | 280 pm |
| C - N | 133 pm | C - N | 148 pm | N-H --- O=C | 290 pm |
| N - Ca | 146 pm | C - O | 143 pm | O-H --- O=C | 280 pm |
[edit] Primary structure
The sequence of the different amino acids is called the primary structure of the peptide or protein. Counting of residues always starts at the N-terminal end (NH2-group), which is the end where the amino group is not involved in a peptide bond. The primary structure of a protein is determined by the gene corresponding to the protein. A specific sequence of nucleotides in DNA is transcribed into mRNA, which is read by the ribosome in a process called translation. The sequence of a protein is unique to that protein, and defines the structure and function of the protein. The sequence of a protein can be determined by methods such as Edman degradation or tandem mass spectrometry. Often however, it is read directly from the sequence of the gene using the genetic code. Post-transcriptional modifications such as disulfide formation, phosphorylations and glycosylations are usually also considered a part of the primary structure, and cannot be read from the gene.
[edit] Secondary structure
By building models of peptides using known information about bond lengths and angles, the first elements of secondary structure, the alpha helix and the beta sheet, were suggested in 1951 by Linus Pauling and coworkers.[2] Both the alpha helix and the beta-sheet represent a way of saturating all the hydrogen bond donors and acceptors in the peptide backbone. These secondary structure elements only depend on properties that all the residues have in common, explaining why they occur frequently in most proteins. Since then other elements of secondary structure have been discovered such as various loops and other forms of helices. The part of the backbone that is not in a regular secondary structure is said to be random coil. Each of these two secondary structure elements have a regular geometry, meaning they are constrained to specific values of the dihedral angles ψ and φ. Thus they can be found in a specific region of the Ramachandran plot.


Backbone
|

Secondary structure cartoon ("ribbon" or "linguini diagram")
|
Turns, loops and a few other secondary structure elements such as a 3-10 helix complete the picture. We have now enough pieces to assemble a complete protein, displaying its typical tertiary structure.
[edit] Tertiary structure
The elements of secondary structure are usually folded into a compact shape using a variety of loops and turns. The formation of tertiary structure is usually driven by the burial of hydrophobic residues, but other interactions such as hydrogen bonding, ionic interactions and disulfide bonds can also stabilize the tertiary structure. The tertiary structure encompasses all the noncovalent interactions that are not considered secondary structure, and is what defines the overall fold of the protein, and is usually indispensable for the function of the protein.
[edit] Quaternary structure
The quaternary structure is the interaction between several chains of peptide bonds. The individual chains are called subunits. The individual subunits are usually not covalently connected, but might be connected by a disulfide bond. Not all proteins have quaternary structure, since they might be functional as monomers. The quaternary structure is stabilized by the same range of interactions as the tertiary structure. Complexes of two or more polypeptides (i.e. multiple subunits) are called multimers. Specifically it would be called a dimer if it contains two subunits, a trimer if it contains three subunits, and a tetramer if it contains four subunits. The subunits are usually related to one another by symmetry axes, such as a 2-fold axis in a dimer. Multimers made up of identical subunits may be referred to with a prefix of "homo-" (e.g. a homotetramer) and those made up of different subunits may be referred to with a prefix of "hetero-" (e.g. a heterotetramer, such as the two alpha and two beta chains of hemoglobin).
[edit] Side chain conformation
The atoms along the side chain are named with Greek letters in Greek alphabetical order: α, β, γ, δ, є and so on. Cα refers to the carbon atom closest to the carbonyl group of that amino acid, Cβ the second closest and so on. The Cα is usually considered a part of the backbone. The dihedral angles around the bonds between these atoms are named χ1, χ2, χ3 etc. E.g. the first and second carbon atom in the side chain of lysine is named α and β, and the dihedral angle around the α-β bond is named χ1. Side chains can be in different conformations called gauche(-), trans and gauche(+). Side chains generally tend to try to come into a staggered conformation around χ2, driven by the minimization of the overlap between the electron orbitals of the hydrogen atoms.
[edit] Domains, motifs, and folds in protein structure
Many proteins are organized into several units. A structural domain is an element of the protein's overall structure that is self-stabilizing and often folds independently of the rest of the protein chain. Many domains are not unique to the protein products of one gene or one gene family but instead appear in a variety of proteins. Domains often are named and singled out because they figure prominently in the biological function of the protein they belong to; for example, the "calcium-binding domain of calmodulin". Because they are self-stabilizing, domains can be "swapped" by genetic engineering between one protein and another to make chimeras. A motif in this sense refers to a small specific combination of secondary structural elements (such as helix-turn-helix). These elements are often called supersecondary structures. Fold refers to a global type of arrangement, like helix bundle or beta-barrel. Structure motifs usually consist of just a few elements, e.g. the 'helix-turn-helix' has just three. Note that while the spatial sequence of elements is the same in all instances of a motif, they may be encoded in any order within the underlying gene. Protein structural motifs often include loops of variable length and unspecified structure, which in effect create the "slack" necessary to bring together in space two elements that are not encoded by immediately adjacent DNA sequences in a gene. Note also that even when two genes encode secondary structural elements of a motif in the same order, nevertheless they may specify somewhat different sequences of amino acids. This is true not only because of the complicated relationship between tertiary and primary structure, but because the size of the elements varies from one protein and the next. Despite the fact that there are about 100,000 different proteins expressed in eukaryotic systems, there are much fewer different domains, structural motifs and folds. This is partly a consequence of evolution, since genes or parts of genes can be doubled or moved around within the genome. This means that, for example, a protein domain might be moved from one protein to another thus giving the protein a new function. Because of these mechanisms pathways and mechanisms tends to be reused in several different proteins.
[edit] Protein folding
The process by which the higher structures form is called protein folding and is a consequence of the primary structure. A unique polypeptide may have more than one stable folded conformation, which could have a different biological activity, but usually, only one conformation is considered to be the active, or native conformation.
[edit] Structure classification
Several methods have been developed for the structural classification of proteins. These seek to classify the data in the Protein Data Bank in a structured order. Several databases exist which classify proteins using different methods. SCOP, CATH and FSSP are the largest ones. The methods used are purely manual, manual and automated, and purely automated. Work is being done to better integrate the current data. The classification is consistent between SCOP, CATH and FSSP for the majority of proteins which have been classified, but there are still some differences and inconsistencies.
[edit] Protein structure determination
Around 90% of the protein structures available in the Protein Data Bank have been determined by X-ray crystallography. This method allows one to measure the 3D density distribution of electrons in the protein (in the crystallized state) and thereby infer the 3D coordinates of all the atoms to be determined to a certain resolution. Roughly 9% of the known protein structures have been obtained by Nuclear Magnetic Resonance techniques, which can also be used to determine secondary structure. Note that aspects of the secondary structure as whole can be determined via other biochemical techniques such as circular dichroism. Secondary structure can also be predicted with a high degree of accuracy (see next section). Cryo-electron microscopy has recently become a means of determining protein structures to high resolution (less than 5 angstroms or 0.5 nanometer) and is anticipated to increase in power as a tool for high resolution work in the next decade. This technique is still a valuable resource for researchers working with very large protein complexes such as virus coat proteins and amyloid fibers.
| Resolution | Meaning |
| >4.0 | Individual coordinates meaningless |
| 3.0 - 4.0 | Fold possibly correct, but errors are very likely. Many sidechains placed with wrong rotamer. |
| 2.5 - 3.0 | Fold likely correct except that some surface loops might be mismodelled. Several long, thin sidechains (lys, glu, gln, etc) and small sidechains (ser, val, thr, etc) likely to have wrong rotamers. |
| 2.0 - 2.5 | As 2.5 - 3.0, but number of sidechains in wrong rotamer is considerably less. Many small errors can normally be detected. Fold normally correct and number of errors in surface loops is small. Water molecules and small ligands become visible. |
| 1.5 - 2.0 | Few residues have wrong rotamer. Many small errors can normally be detected. Folds are extremely rarely incorrect, even in surface loops. |
| 0.5 - 1.5 | In general, structures have almost no errors at this resolution. Rotamer libraries and geometry studies are made from these structures. |
[edit] Computational prediction of protein structure
The generation of a protein sequence is much simpler than the generation of a protein structure. However, the structure of a protein gives much more insight in the function of the protein than its sequence. Therefore, a number of methods for the computational prediction of protein structure from its sequence have been proposed. Ab initio prediction methods use just the sequence of the protein. Threading uses existing protein structures. Homology Modeling to build a reliable 3D model for a protein of unknown structure from one or more related proteins of known structure. The recent progress and challenges in protein structure prediction was reviewed by Zhang [3].
Rosetta@home is a distributed computing project which tries to predict the structures of proteins with massive sampling on thousands of home computers. Foldit is a video game designed to use human pattern recognition and puzzle solving abilities to improve existing software.
[edit] Software
There are many available software packages, such as free web-based STING, used to visualize and analyze protein structures. Another example is the FeatureMap3D web-server which can visualize the quality of a protein-protein alignment in 3D and be used to map sequence feature annotation such as the underlying Intron/Exon structure onto a protein structure.
Several packages, such as Quantum Pharmaceuticals software[4], can be used to predict conformational changes of proteins and its influence on protein's functions.
Several methods have been developed to compare structures of different proteins. Please see structural alignment.
Computational tools are also frequently employed to check experimental and theoretical models of protein structures for errors (examples: ProSA, NQ-Flipper, Verify3D, ANOLEA, WHAT_CHECK).
Software for molecular mechanics modeling useful for building and simulation of protein models.
[edit] References
- ^ Brocchieri L, Karlin S (2005-06-10). Protein length in eukaryotic and prokaryotic proteomes. 33. p. 3390-3400. doi:. PMID 15951512.
- ^ Pauling L, Corey RB, Branson HR (1951). "The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain". Proc Natl Acad Sci USA 37 (4): 205-211. PMID 14816373.
- ^ Zhang Y (2008). "Progress and challenges in protein structure prediction". Curr Opin Struct Biol 18 (3): 342-348. doi:. Entrez Pubmed 18436442. PMID 18436442.
- ^ Quantum Pharmaceuticals software
[edit] Further reading
- Chiang YS, Gelfand TI, Kister AE, Gelfand IM (2007). "New classification of supersecondary structures of sandwich-like proteins uncovers strict patterns of strand assemblage.". Proteins. 68 (4): 915-921. PMID 17557333. http://www.ncbi.nlm.nih.gov/pubmed/17557333.
- Habeck M, Nilges M, Rieping W (2005). "Bayesian inference applied to macromolecular structure determination". Physical review. E, Statistical, nonlinear, and soft matter physics 72 (3 Pt 1): 031912. PMID 16241487. http://www.spineurope.org/publications/Habeck%20et%20al%20031912%202005.pdf. (Bayesian computational methods for the structure determination from NMR data)
[edit] External links
- SSS Database super-secondary structure protein database
- SPROUTS (Structural Prediction for pRotein fOlding Utility System)
- ProSA-web Web service for the recognition of errors in experimentally or theoretically determined protein structures
- NQ-Flipper Check for unfavorable rotamers of Asn and Gln residues in protein structures
- servers That check nearly 200 aspects of protein structure, like packing, geometry, unfavourable rotamers in general of for Asn, Gln and His especially, strange water molecules, backbone conformations, atom nomenclature, symmetry parameters, etc.
- Bioinformatics course. An interactive, fully free, course explaining many of the aspects discussed in this wiki entry.
|
|||||||||||
|
|||||||||||||||||||||||||||||

